Command Palette
Search for a command to run...
Auf Der Grundlage Von 14.000 Realen Datensätzen Schlugen Die University of Washington/Microsoft Und Andere GigaTIME Vor, Um Einen Panoramaatlas Der Immunologischen Mikroumgebung Von Tumoren Zu erstellen.
Im evolutionären Kontext von Krebs dominiert die Tumormikroumgebung nicht nur Wachstum, Invasion und Metastasierung von Krebszellen, sondern beeinflusst auch maßgeblich das Ansprechen auf die Therapie und die endgültige Prognose des Patienten. Es handelt sich dabei nicht um ein isoliertes Vorgehen der Krebszellen, sondern um ein hochdynamisches Ökosystem: Immunzellen, Fibroblasten, Endothelzellen und andere Zellen interagieren miteinander und sind in die extrazelluläre Matrix eingebettet, deren Struktur und Funktion verändert wurden. So entsteht ein präzises und komplexes pathologisches Netzwerk.
Der Schlüssel zur Entschlüsselung dieses Netzwerks liegt im Verständnis der Funktionszustände und Wechselwirkungen der Zellen, wobei die Aktivierungsniveaus spezifischer Proteine entscheidende „molekulare Codes“ darstellen. TraditionellDie Immunhistochemie (IHC) hat sich aufgrund ihrer Fähigkeit, die Proteinlokalisierung visuell darzustellen, zu einem klassischen Werkzeug zur Entschlüsselung von Codes entwickelt.Die PD-L1-Färbung wird beispielsweise häufig eingesetzt, um den Status von Immun-Checkpoints zu bestimmen und so die Wirksamkeit von Immuntherapien vorherzusagen. Allerdings kann die Immunhistochemie (IHC) jeweils nur Informationen über ein Protein erfassen, was die Rekonstruktion der tatsächlichen Wechselwirkung mehrerer Proteine erschwert. Dies stellt ein wesentliches Hindernis für ein tieferes Verständnis des Dialogmechanismus zwischen Tumorzellen und Immunzellen dar.
Um diese Einschränkung zu überwinden, wurde die Multiplex-Immunfluoreszenz-Technologie (mIF) entwickelt. Sie ermöglicht die gleichzeitige Darstellung der räumlichen Verteilung mehrerer Proteine auf einem einzigen Gewebeschnitt und erhält dabei vollständig die Kontextinformationen der Gewebestruktur.Diese Technologie ist jedoch teuer und mit einem komplizierten Verfahren verbunden, da Färben, Bildgebung und Analyse extrem zeitaufwändig sind.Dies erschwert die Erhebung umfangreicher Daten und behindert die klinische Umsetzung.
Im Gegensatz dazu sind H&E-gefärbte Schnitte in der klinischen Praxis weit verbreitet und kostengünstig. Obwohl sie die Proteinaktivität nicht direkt sichtbar machen können, erhalten sie die Gesamtstruktur des Gewebes und die Details der Zellmorphologie vollständig. Die darin verborgenen Merkmale spiegeln möglicherweise indirekt den Funktionszustand der Zellen wider, doch diese subtilen und komplexen Muster übersteigen oft die Grenzen der menschlichen Wahrnehmung.
In den letzten Jahren haben Durchbrüche in der Technologie der künstlichen Intelligenz (KI) neue Möglichkeiten eröffnet. Durch das Vortraining mit riesigen Mengen pathologischer Bilder hat die KI ihre leistungsstarken Fähigkeiten in der visuellen Analyse und Merkmalsextraktion unter Beweis gestellt. Daraus ergibt sich eine zentrale Frage: Kann KI genutzt werden, um Informationen zur Proteinaktivierung aus leicht verfügbaren H&E-Bildern zu „entschlüsseln“ – Informationen, deren Erfassung bisher teure mIF-Technologie erforderte?
Ausgehend von dieser Denkweise,Ein Forschungsteam bestehend aus Microsoft Research, der University of Washington und Providence Genomics hat GigaTIME, ein multimodales Framework für künstliche Intelligenz, vorgeschlagen.Mithilfe fortschrittlicher multimodaler Lerntechnologie lassen sich virtuelle mIF-Karten aus herkömmlichen H&E-gefärbten Gewebeschnitten generieren. Das Forschungsteam wandte diese Technologie auf eine Kohorte von über 14.000 Krebspatienten am Providence Medical Center in den USA an. Die Patienten umfassten 24 Krebsarten und 306 Subtypen und generierten schließlich fast 300.000 virtuelle mIF-Bilder. Dadurch gelang eine systematische Modellierung der Tumor-Immunmikroumgebung in einer großen und heterogenen Population.
Die zugehörigen Forschungsergebnisse mit dem Titel „Multimodale KI generiert virtuelle Population für die Modellierung der Tumormikroumgebung“ wurden in Cell veröffentlicht.
Forschungshighlights:
* GigaTIME nutzt multimodale KI, um H&E-Pathologiepräparate in räumliche Proteomikdaten umzuwandeln und virtuelle Populationen zu generieren, die Zellzustände aus routinemäßigen H&E-Präparaten enthalten.
* Unterstützt die klinische Forschung im großen Maßstab und die Patientenstratifizierung und deckt neuartige räumliche und kombinatorische Proteinaktivierungsmuster auf.

Papieradresse:https://www.cell.com/cell/fulltext/S0092-8674(25)01312-1
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „Mehrfachimpfungen“, um das vollständige PDF zu erhalten.
Weitere Artikel zu den Grenzen der KI:
https://hyper.ai/papers
Datensätze: Aufbau eines vollständig geschlossenen Kreislaufs von der Schulung bis zur Anwendung
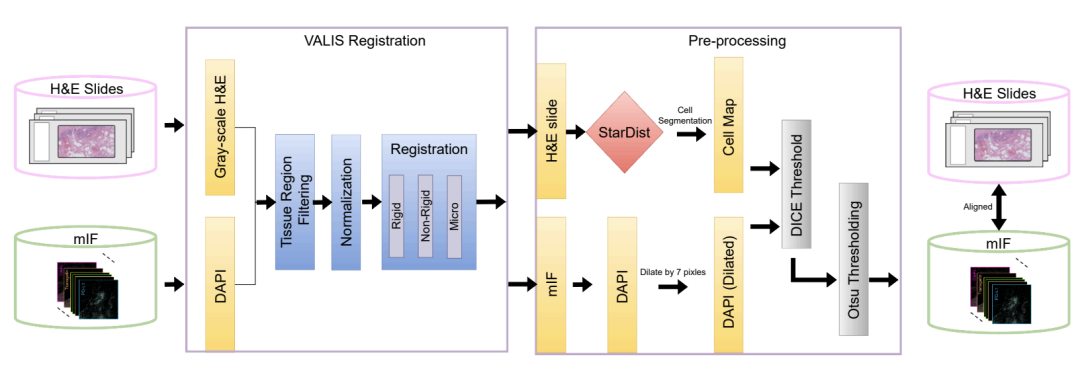
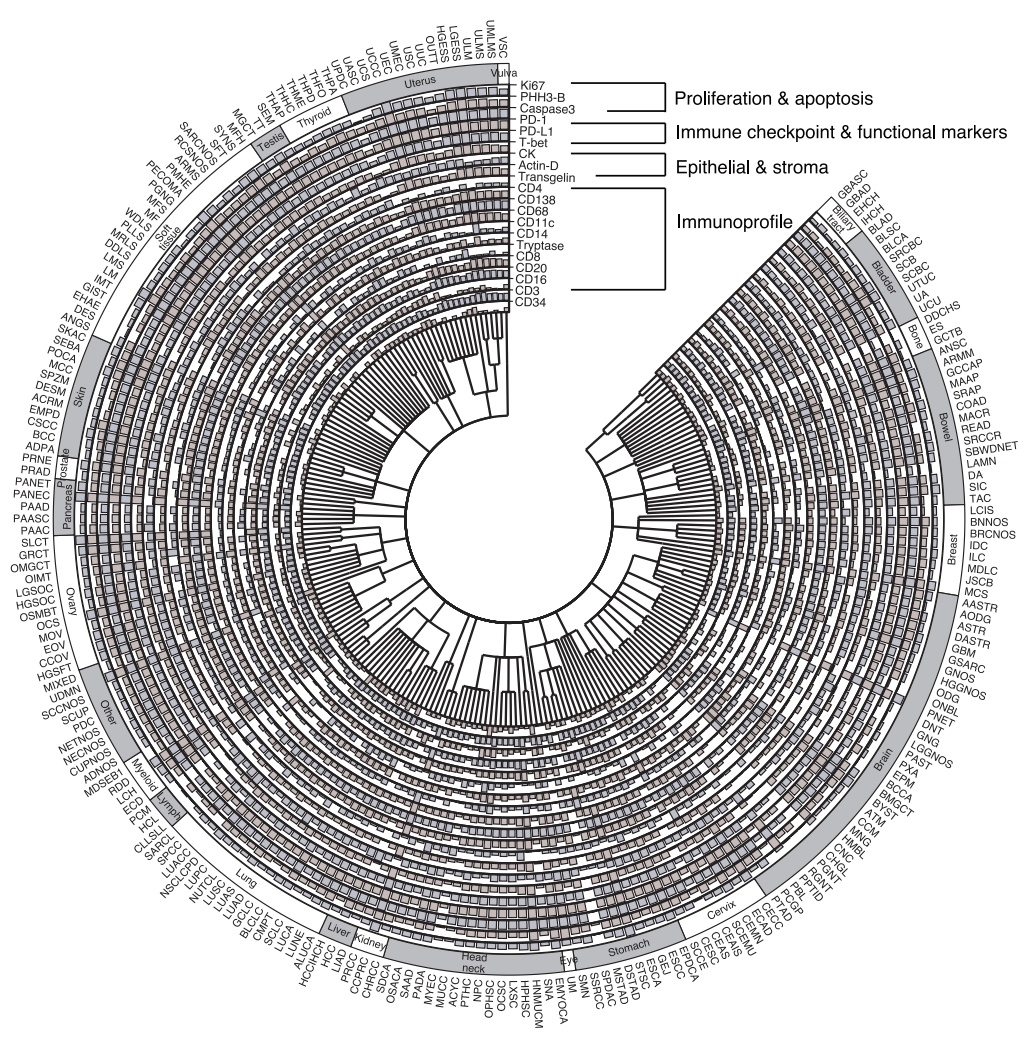
Das Training eines Modells erfordert zunächst die Auflösung eines grundlegenden Widerspruchs: Die H&E-Färbung, die in der klinischen Praxis weit verbreitet und kostengünstig ist, kann die Proteinaktivität nicht direkt aufzeigen, während die mIF-Technologie, die räumliche Beziehungen zwischen mehreren Proteinen sichtbar macht, teuer und komplex ist, was eine großflächige Anwendung erschwert. Um ein KI-Modell zu entwickeln, das diese beiden Bildgebungsverfahren miteinander verbindet,Das Forschungsteam nutzte die COMET-Plattform, um 441 mIF-Bilder von 21 H&E-gefärbten Schnitten zu sammeln.Wie in der Abbildung unten dargestellt, umfassen diese Bilder insgesamt 21 wichtige Biomarker, von Kernproteinen wie DAPI und PHH3 über Oberflächenproteine wie CD4 und CD11c bis hin zu zytoplasmatischen Proteinen wie CD68. Sie liefern wichtige Erkenntnisse für die Analyse der Zusammensetzung, des Funktionszustands und der Aktivität von Immunzellen in der Tumormikroumgebung.

Nachdem die Bildpaare erfasst wurden, besteht die größere Herausforderung darin, daraus hochwertige Trainingsdaten zu extrahieren. Zu diesem Zweck entwickelte das Team, wie in der Abbildung unten dargestellt, einen sorgfältigen Verarbeitungsablauf: Zunächst werden das H&E-Bild und das mIF-Bild mithilfe des VALIS-Tools pixelgenau ausgerichtet. Anschließend werden mit dem StarDist-Algorithmus die einzelnen Zellen im Bild identifiziert und segmentiert. Schließlich wird anhand des Dice-Koeffizienten der Bildbereich mit der höchsten Registrierungsqualität ausgewählt.
Durch mehrere Ebenen der QualitätskontrolleDas Team wählte aus den ursprünglichen Daten mit 40 Millionen Zellen 10 Millionen qualitativ hochwertige Zellen aus und teilte diese in einen Trainingsdatensatz, einen Validierungsdatensatz und einen unabhängigen Testdatensatz auf.Zusätzlich wurden in der Studie Brust- und Hirntumorproben aus Gewebemikroarrays als externer Validierungsdatensatz verwendet. Diese Proben unterschieden sich hinsichtlich Gewebestruktur und -morphologie deutlich von den Trainingsdaten – sie erschienen als kleine zylindrische Gewebeblöcke, die durch leere Bereiche voneinander getrennt waren, anstatt als große, zusammenhängende Gewebeschnitte in den Trainingsdaten. Dadurch wurde die Generalisierungsfähigkeit des Modells im Umgang mit neuen Probentypen und unbekannten Krebsarten effektiv getestet.
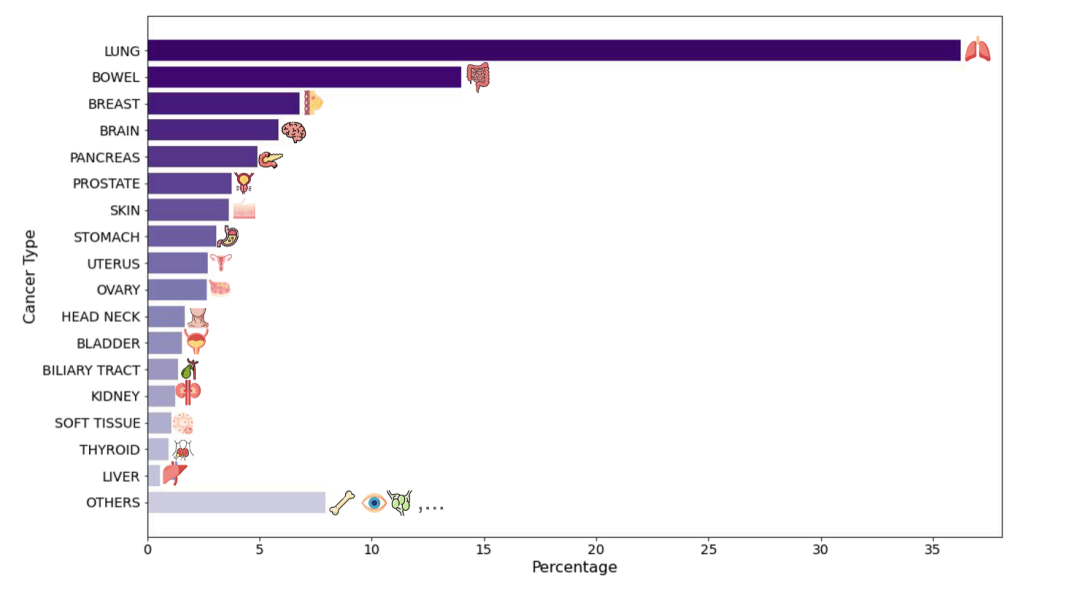
Auf Ebene der Modellanwendung wurden im Rahmen der Studie zwei große und sich ergänzende virtuelle Bevölkerungskohorten erstellt.Die erste Kohorte stammt aus dem klinischen Netzwerk von Providence Health, einem US-amerikanischen Gesundheitskonzern, und umfasst H&E-gefärbte Gewebeschnitte von 14.256 Krebspatienten aus 51 Krankenhäusern und über 1.000 Kliniken. Sie deckt 24 Hauptkrebsarten und 306 Subtypen ab und integriert umfangreiche klinische Informationen wie genomische Biomarker, pathologisches Stadium und Überlebensdaten. Der besondere Wert dieses Datensatzes liegt in seiner Realitätsnähe: eine äußerst diverse Patientenpopulation, die das gesamte Spektrum der Krankheitsstadien von früh bis spät abdeckt und somit die Komplexität der klinischen Praxis realistisch widerspiegelt.
Die zweite Kohorte wurde der öffentlichen Datenbank des Cancer Genome Atlas (TCGA) entnommen.Die Studie umfasste 10.200 H&E-Präparate, vorwiegend aus unbehandelten Fällen im Frühstadium nach chirurgischen Eingriffen. Diese beiden Patientengruppen unterschieden sich deutlich hinsichtlich Patientenherkunft, Krankheitsstadium und klinischem Kontext. Dieses differenzierte Studiendesign bot hervorragende Voraussetzungen für die Validierung der Zuverlässigkeit und Generalisierbarkeit des Modells: Konsistente und robuste biologische Schlussfolgerungen aus solch unterschiedlichen Datensätzen deuten stark auf sein breites klinisches Potenzial hin.
GigaTIME: Intelligente Brücken zwischen Form und Funktion bauen
Das GigaTIME-Modell adressiert direkt einen zentralen Engpass in der Erforschung der Tumorimmunmikroumgebung: Die kostenintensive und wenig effiziente mIF-Technologie ist schwer zu verbreiten, während routinemäßige klinische H&E-Färbungen die funktionelle Aktivität von Proteinen nicht direkt widerspiegeln. Dieses Modell nutzt künstliche Intelligenz, um aus H&E-Bildern virtuelle mIF-Bilder zu generieren und bietet so einen praktikablen Weg für eine kostengünstige, systematische Analyse der Tumorimmunmikroumgebung im Bevölkerungsmaßstab.
Das Modell verwendet ein sorgfältig entworfenes Patchwork-Encoder-Decoder-Framework, dessen Kern auf einem verschachtelten U-förmigen Netzwerk basiert.Der Vorteil dieser Architektur liegt in ihrer Fähigkeit, gleichzeitig sowohl subtile lokale Merkmale als auch die globale Organisationsstruktur eines Bildes zu erfassen. Konkret extrahiert der Encoder des Netzwerks mehrstufige Merkmalsdarstellungen aus dem 256×256 Pixel großen H&E-Bildausschnitt mittels Faltung und Downsampling. Der Decoder rekonstruiert diese abstrakten Merkmale anschließend durch Upsampling und Merkmalsfusion zu einem virtuellen mIF-Bild mit räumlicher Auflösung. Dieses Design ermöglicht es dem Modell, sich sowohl auf die Feinmorphologie einzelner Zellen als auch auf die Organisationsmuster von Zellpopulationen zu konzentrieren.
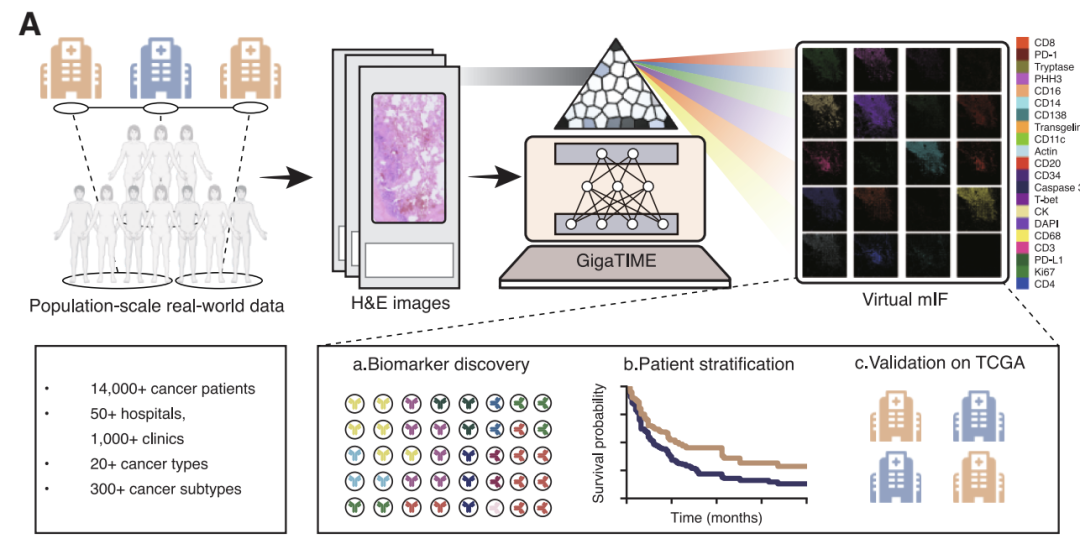
Auf der Output-Ebene spiegelt das Design des Modells eine tiefgreifende Auseinandersetzung mit biologischen Fragestellungen wider.Für jeden der 21 voreingestellten Proteinkanäle führt GigaTIME eine binäre Klassifizierungsvorhersage für jedes Pixel im Eingabebild durch.Das System ermittelt, ob ein bestimmtes Protein an einer gegebenen Stelle aktiviert ist, und erstellt so eine Proteinaktivitätskarte auf Pixelebene. Diese lokalen Vorhersagen lassen sich nahtlos zu einem virtuellen mIF-Bild des gesamten Gewebeschnitts zusammenfügen. Dies ermöglicht die Berechnung verschiedener quantitativer Indikatoren, wie z. B. der Aktivierungsdichte und des räumlichen Verteilungsmusters spezifischer Proteine im Tumorbereich, und liefert eine solide Datengrundlage für nachfolgende Hochdurchsatzanalysen und klinische Korrelationsstudien.
Um ein effektives Modelllernen zu gewährleisten, wurde die Trainingsstrategie systematisch optimiert.Die Verlustfunktion kombiniert geschickt den Dice-Koeffizienten und die binäre Kreuzentropie: Ersterer optimiert die räumliche Kontur des vorhergesagten aktiven Bereichs im Vergleich zum tatsächlichen Bereich, während letztere die Klassifizierungsgenauigkeit jedes einzelnen Pixels verbessert. Das Zusammenspiel beider Funktionen gewährleistet sowohl eine präzise Rekonstruktion des globalen räumlichen Musters als auch Zuverlässigkeit im Detail. Das Modell wurde über 250 Epochen auf 8 NVIDIA A100 GPUs mit einer Batchgröße von 16 und einer Lernrate von 0,0001 trainiert. Alle wichtigen Hyperparameter wurden anhand der Ergebnisse des Validierungsdatensatzes durch System-Debugging ermittelt.
Es ist besonders wichtig zu betonen, dass der Erfolg des Modells stark von qualitativ hochwertigen Trainingsdaten abhängt.Das Forschungsteam wandte strenge Verfahren zur Bildregistrierung, Zellsegmentierung und Qualitätskontrolle an.Aus den massiven Ausgangsdaten wurden 10 Millionen qualitativ hochwertige Zellen für das Training ausgewählt, um sicherzustellen, dass das Modell robuste, zuverlässige und biologisch sinnvolle multimodale Zuordnungen lernt und nicht nur oberflächliche statistische Regelmäßigkeiten oder verrauschte Muster.
Großangelegte Erkenntnisse basierend auf fast 300.000 virtuellen Bildern: GigaTIME deckt 1.234 klinische Zusammenhänge auf
Um die Leistungsfähigkeit und den Wert von GigaTIME umfassend zu bewerten, entwickelte das Forschungsteam ein systematisches Bewertungsschema, das auf zwei Dimensionen basiert: Technologievalidierung und klinische Ergebnisse.
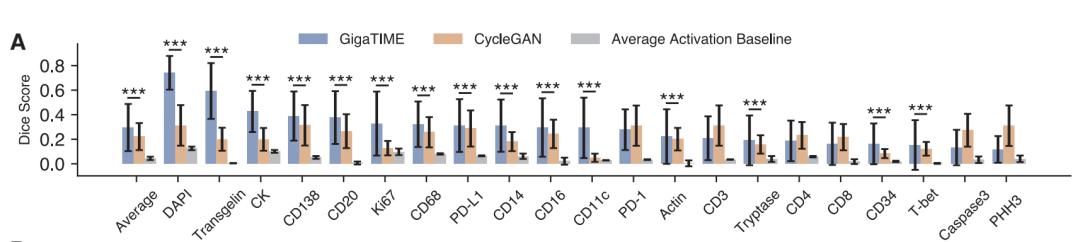
Im Hinblick auf die technische Überprüfung,Die Studie bewertete die Bildkonvertierungsfähigkeiten des Modells auf drei Ebenen: Pixel, Zelle und Schicht.Auf Pixelebene übertrifft GigaTIME das Basismodell CycleGAN in 15 von 21 Proteinkanälen deutlich. Beispielsweise erreicht GigaTIME im DAPI-Kanal einen Dice-Koeffizienten von 0,72, der den Wert von 0,12 des einfachen statistischen Basismodells deutlich übertrifft.
Auf zellulärer EbeneGigaTIME erreichte auf dem DAPI-Kanal eine Korrelation von 0,59, während CycleGAN nur 0,03 erreichte und sich damit einem Zufallsniveau annäherte.
Auf der Ebene der SchichtDer DAPI-Kanalkorrelationskoeffizient von GigaTIME erreicht einen Wert von 0,98, mit einem Durchschnitt von 0,56 über alle Kanäle hinweg, während der Wert von CycleGAN nahe bei 0 liegt. Diese Ergebnisse zeigen, dass überwachtes Training auf Basis hochwertiger gepaarter Daten für eine präzise intermodale Konvertierung unerlässlich ist.
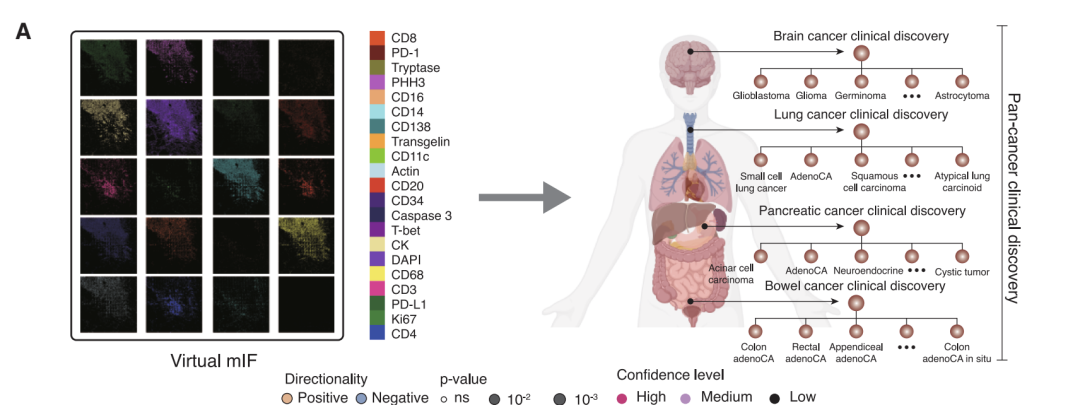
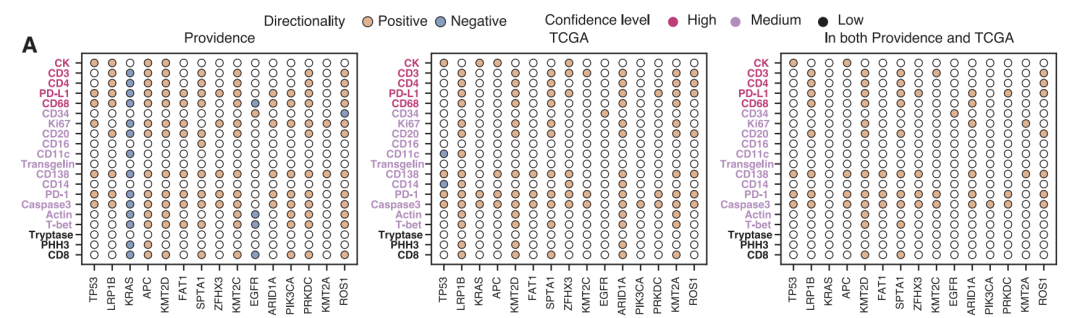
Hinsichtlich der klinischen Befunde wurden in der Studie knapp 300.000 virtuelle mIF-Bilder von 14.256 Patienten verwendet.Der Zusammenhang zwischen virtueller Proteinexpression und 20 klinischen Biomarkern wurde systematisch analysiert.Nach strengen statistischen Tests und zahlreichen Korrekturen wurden insgesamt 1.234 signifikante Assoziationen identifiziert, die sich auf drei Ebenen verteilen: Pan-Krebs, Krebsart und Krebssubtyp.
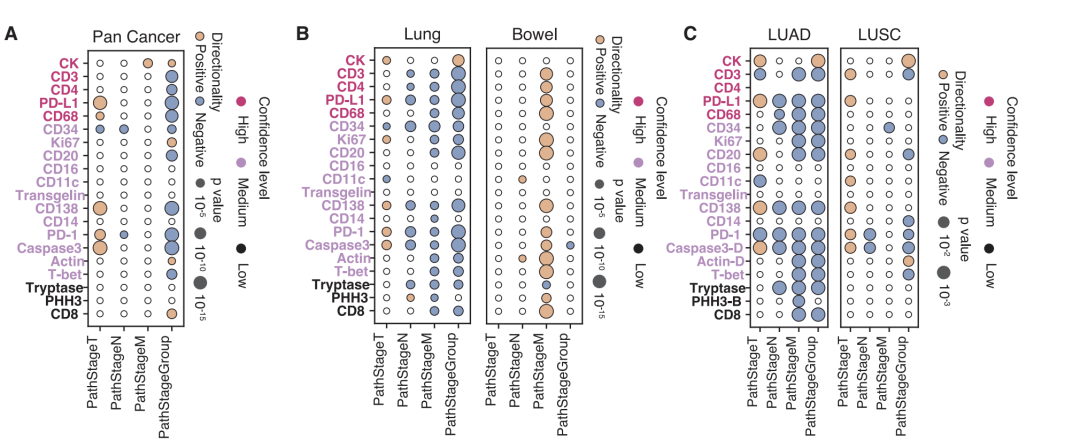
Unter den 175 in der Pan-Cancer-Analyse identifizierten Assoziationen waren eine hohe Mutationslast des Tumors und eine hohe Mikrosatelliteninstabilität signifikant mit einer verstärkten Aktivierung mehrerer Immuninfiltrationsmarker (CD138, CD20, CD68 und CD4) assoziiert, was mit einem antigengetriebenen Immunaktivierungsmechanismus übereinstimmt. Es wurden auch neue Hinweise gefunden: KMT2D-Mutationen zeigten eine starke positive Korrelation mit Immunmarkern, was auf eine potenzielle Förderung der Immuninfiltration hindeutet; KRAS-Mutationen hingegen zeigten eine negative Korrelation, was einen Immunabwehrphänotyp widerspiegelt. In spezifischen Krebsarten und -subtypen deckte das Modell zahlreiche spezifische Assoziationen auf. Beispielsweise wurde die starke Korrelation zwischen T-bet- und TP53-Mutationen bei Hirntumoren auf Pan-Cancer-Ebene nicht festgestellt, was möglicherweise mit der einzigartigen Immunmikroumgebung des zentralen Nervensystems zusammenhängt. Die Subtypanalyse von Lungenkrebs zeigte, dass PRKDC-Mutationen im Lungenadenokarzinom stärker mit Immunantwortmarkern assoziiert waren als im Plattenepithelkarzinom, was die Bedeutung der Interpretation der Daten im Zusammenhang mit dem histologischen Kontext bestätigt.
Die Studie bestätigte auch den Wert virtueller Daten für klinische Ergebnisse.Die Analyse ergab eine positive Korrelation zwischen der Größe des Primärtumors (T-Stadium) und Immun-Checkpoints sowie Invasionsmarkern. Dieser Zusammenhang kehrte sich jedoch in fortgeschrittenen Stadien um, was darauf hindeutet, dass fortgeschrittene Tumoren primär durch andere Mechanismen der Immunflucht bedingt sein könnten. In der Überlebensanalyse erwiesen sich die kombinierten Merkmale, die alle 21 Signalwege integrierten, hinsichtlich der Patientenstratifizierung als überlegen gegenüber Einzelproteinanalysen. Dies unterstreicht den Wert der kombinierten Analyse mehrerer Parameter.
Um die Zuverlässigkeit zu gewährleisten, wurden alle zentralen Ergebnisse in unabhängigen Kohorten aus TCGA validiert. Trotz signifikanter Unterschiede in Herkunft und klinischen Merkmalen zwischen den beiden Populationen blieben die Kernergebnisse hochgradig konsistent (Spearman-Korrelationskoeffizient von 0,88 auf Ebene der Krebs-Subtypen).Die 80 häufig identifizierten signifikanten Assoziationen zeigten eine extrem hohe statistische Anreicherung (p<2×10⁻⁹).Die virtuelle Population von Providence Health wies unterdessen 331 signifikante Assoziationen mehr auf der Ebene aller Krebsarten auf als TCGA, was den einzigartigen Wert von groß angelegten Daten aus der realen Welt unterstreicht.
Explorative Analysen zeigten zudem den Wert komplexer räumlicher Muster auf. Indikatoren wie Entropie, Signal-Rausch-Verhältnis und Schärfe waren der einfachen Aktivierungsdichte in 89, 63 bzw. 79 Protein-Biomarker-Paaren überlegen. Die Studie entdeckte außerdem synergistische Effekte zwischen Proteinen:Die Kombination von CD138 und CD68 war bei der Vorhersage von 20 Biomarkern den einzelnen Proteinen überlegen.Dreizehn dieser Unterschiede waren statistisch signifikant, was darauf hindeutet, dass Plasmazellen und Makrophagen möglicherweise über einen antikörpervermittelten Mechanismus zusammenarbeiten, um Tumore zu bekämpfen.
KI-gestützt: Von virtuellen Proteinkarten zu neuen Horizonten in der Krebsforschung
Die Generierung virtueller Proteomikbilder aus routinemäßigen Pathologiepräparaten mithilfe von KI ist ein zentraler Innovationsansatz in der digitalen Pathologie und der Computerbiologie. Diese Richtung hat weltweit das Interesse führender akademischer Einrichtungen geweckt und auch die Kommerzialisierung durch Biotechnologieunternehmen vorangetrieben.
In der WissenschaftDas von der Stanford University in Nature Medicine veröffentlichte HEX-ModellDas System, trainiert mit 819.000 gepaarten Bildblöcken, kann die räumliche Expression von 40 Biomarkern vorhersagen und bietet damit eine umfassendere Proteinabdeckung als GigaTIME. Das von der University of California, San Francisco in Science Translational Medicine veröffentlichte DeepHeme-System, basierend auf fast 50.000 hochwertigen multizentrischen Datensätzen, hat eine präzise Klassifizierung von 23 Knochenmarkzelltypen erreicht und liefert damit ein Paradigma für die Automatisierung der hämatologischen Diagnostik.
In der Branche wird Reveal Biosciences von der Bill & Melinda Gates Foundation unterstützt.Entwicklung einer Plattform zur Extraktion „digitaler Biomarker“ aus pathologischen Bildern.Die globale Gesundheitsforschung wird beschleunigt. Ein weiterer Ansatz besteht darin, Kosten durch Hardware-Innovationen zu senken, beispielsweise durch die mikrofluidischen Geräte von Micronit, die den Verbrauch von Proben und Reagenzien deutlich reduzieren. Die FDA-zugelassene Diagnostikplattform für Lungenknoten von Optellum bietet ein kommerzielles Paradigma und einen regulatorischen Präzedenzfall für die Gewinnung detaillierterer Informationen aus Routinedaten zur klinischen Entscheidungsfindung.
GigaTIME ist ein bedeutender Meilenstein auf diesem Gebiet.Dies demonstriert nicht nur das enorme Potenzial multimodaler KI bei der Untersuchung der immunologischen Mikroumgebung von Tumoren, sondern bietet auch einen wiederverwendbaren technischen Rahmen und Datenressourcen für nachfolgende Forschungen.Die zukünftige Entwicklung wird von der kombinierten Weiterentwicklung der Datengenerierungsfähigkeiten mittels „Virtual Reality“ und kostengünstiger Detektionstechnologien abhängen, um letztendlich transformative Werkzeuge zum Verständnis der Komplexität von Tumoren und zur Beschleunigung der Präzisionsmedizin bereitzustellen.
Referenzlinks:
1.https://mp.weixin.qq.com/s/AsqSemP3idCbIJ7xQ3gXGg
2.https://mp.weixin.qq.com/s/umg-UrMm6Qe-R-MbLpLZOQ








