Command Palette
Search for a command to run...
Durch Die Integration Von Daten Zu Proteinsequenzen, Dreidimensionalen Strukturen Und Funktionellen Eigenschaften Erstellte Ein Deutsches Team Auf Basis Von Metriklernen Eine „Panoramaansicht“ Der Menschlichen E3-Ubiquitin-Ligasen.
In Organismen ist der zeitgerechte Abbau und die Erneuerung zellulärer Proteine entscheidend für die Aufrechterhaltung der Proteinhomöostase. Das Ubiquitin-Proteasom-System (UPS) ist ein zentraler Mechanismus zur Regulation der Signaltransduktion und des Proteinabbaus. Innerhalb dieses Systems sind E3-Ubiquitin-Ligasen als wichtige katalytische Einheiten für die Erkennung spezifischer Substrate und die Katalyse der Ubiquitinierung verantwortlich und regulieren dadurch Proteinabbau, -lokalisierung und Funktionszustand. Darüber hinaus regulieren E3-Ligasen auch Immun- und Entzündungsprozesse. Aufgrund ihrer gewebespezifischen Expression und ihrer Assoziation mit Entwicklungs- und Stoffwechselsyndromen (einschließlich Krebsentwicklung) haben sich E3-Ligasen zu vielversprechenden Angriffspunkten für Medikamente entwickelt, insbesondere für Zielstrukturen, die zuvor schwer medikamentös angreifbar waren.
Im Vergleich zu den E1- (ca. 10 Typen) und E2-Enzymen (ca. 50 Typen) wurde eine große Anzahl humaner E3-Ligasen (ca. 600 Typen) identifiziert. Dennoch sind viele humane E3-Ligasen nur teilweise charakterisiert, und eine große Anzahl ist noch hypothetisch oder unbekannt. Bis heuteDie untersuchten E3-Ligasen weisen eine hohe Heterogenität auf.Dies macht sie zu einer der vielfältigsten Enzymklassen und stellt einen Engpass für die Mustererkennung und groß angelegte Forschung dar. Daher ist die detaillierte Charakterisierung und Analyse des menschlichen E3-Ligase-Genoms – der Gesamtheit der vom menschlichen Genom kodierten E3-Ligasen – entscheidend für ein umfassendes Verständnis ihrer biologischen Funktionen.
In diesem ZusammenhangEin Forschungsteam der Goethe-Universität in Deutschland hat das „menschliche E3-Ligom“ klassifiziert.Es integriert Daten auf verschiedenen Ebenen, darunter Proteinsequenzen, Domänenzusammensetzung, dreidimensionale Struktur, Funktion und Expressionsmuster.Die Klassifizierungsmethode des Teams basiert auf dem Metrik-Lernparadigma und verwendet einen schwach überwachten hierarchischen Rahmen, um die wahren Beziehungen zwischen der E3-Familie und ihren Unterfamilien zu erfassen.Dieser Ansatz erweitert die traditionelle Klassifizierung der E3-Enzyme (RING-, HECT- und RBR-Klassen), unterscheidet zwischen Multisubunit-Komplexen und monomeren Enzymen und ordnet E3-Enzyme Substraten und potenziellen Wirkstoffzielen zu.
Die zugehörigen Forschungsergebnisse mit dem Titel „Multi-scale classification decodes the complexity of the human E3 ligome“ wurden in Nature Communications veröffentlicht.
Forschungshighlights:
* Die Domänenarchitektur, die dreidimensionale Struktur, die Funktion, das Substratnetzwerk und die Wechselwirkungen kleiner Moleküle bestehender E3-Ligasen werden in einem Klassifizierungsrahmen abgebildet, um allgemeine und familienspezifische Erkenntnisse zu gewinnen.
* Das entwickelte multiskalige Klassifizierungsmodell umfasst sowohl typische als auch atypische E3-Mechanismen und bietet einen vollständigen Leitfaden zum Verständnis der breiten biologischen Landschaft der E3-Ligasen.
* Eröffnet neue Wege für die Entwicklung von Arzneimittelinterventionsstrategien auf der Grundlage von E3-Substratnetzwerken.

Papieradresse:
https://www.nature.com/articles/s41467-025-67450-9
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „E3 Enzym“, um das vollständige PDF zu erhalten.
Weitere Artikel zu den Grenzen der KI:
https://hyper.ai/papers
Datensatz: Erstellung von Daten zur humanen E3-Ubiquitin-Ligase
Das Forschungsteam integrierte zunächst Daten zur menschlichen E3-Ubiquitin-Ligase aus acht unabhängigen Datenquellen.Unter Einbeziehung früherer Literaturberichte und öffentlicher Datenbanken (E3Net, UbiHub, UbiNet 2.0, UniProt, BioGRID usw.) wurde ein vorläufiger Datensatz mit 1448 Proteineinträgen erstellt. Doppelte und potenziell falsch-positive Einträge wurden durch Quervergleich und Konsistenzbewertung der Daten aus verschiedenen Quellen entfernt. Anschließend wurden mithilfe der von InterPro bereitgestellten katalytischen Domänenmerkmale RING, HECT und RBR 462 hochkonfidente katalytische E3-Ubiquitin-Ligasen identifiziert, wodurch das endgültige Genom der humanen E3-Ligasen entstand.
In E3-Komplexen mit mehreren Untereinheiten (wie Cullin-RING-Ligasen) arbeiten drei funktionell unterschiedliche Untereinheiten (Gerüstprotein, Aptamerprotein und Rezeptorprotein) zusammen, um E2~Ub-Moleküle an spezifische Substrate zu binden. Große, starre und zentral gelegene Gerüstproteine (wie die Cullin-Familie, Cul1–Cul5) organisieren den gesamten Ligasekomplex, indem sie gleichzeitig an die Andockstellen der katalytischen RING-Fingerdomäne und des Aptamers/Rezeptors binden. Das Aptamerprotein verbindet die Module, indem es die N-terminale Andockstelle des Gerüstproteins mit den einzelnen Substratrezeptoren verknüpft. Das Rezeptorprotein bestimmt die Substratspezifität, indem es direkt Degradationssignale (Degrons) auf dem Substrat erkennt und bindet, um festzulegen, welche Substrate ubiquitiniert werden (z. B. Skp2, Keap1, VHL).Das Forschungsteam annotierte und kategorisierte unabhängig voneinander drei Untereinheiten: 151 Aptamere, 106 Rezeptoren und 8 Gerüstproteine.Sie nutzten außerdem ihre Protein-Protein-Interaktionen (PPIs), um die Substrate der Multisubunit-E3 zu kartieren.
Anschließend nutzten die Forscher in der Phase des Screenings der katalytischen Domänen die katalytische Fähigkeit als zentrales Kriterium, um Kandidatenproteine streng zu filtern.Mithilfe von Domänendatenbanken wie InterPro identifizierte das System wichtige katalytische Domänen, die in direktem Zusammenhang mit der E3-Aktivität stehen, darunter RING, HECT und RBR.Nur Proteine, die diese Domänen explizit enthalten und ihre Ubiquitin-Ligandenfunktion auf Sequenz- und Strukturebene unterstützen, werden für den Aufbau der finalen „katalytischen E3-Ligase“ beibehalten. Dieser Prozess eliminiert effektiv akzessorische Proteine, die lediglich an der Regulation beteiligt sind, aber keine direkte katalytische Aktivität besitzen, und gewährleistet so die funktionelle Konsistenz des E3-Kernkomplexes.
Ein auf metrischem Lernen basierendes, mehrskaliges Klassifizierungsframework
Um die komplexen Beziehungen innerhalb des menschlichen E3-Ligase-Genoms zu erfassen,Forscher nutzten Methoden des maschinellen Lernens, um eine emergente Distanzmetrik zu erlernen.Das Gesamtkonzept ist im folgenden Diagramm dargestellt:
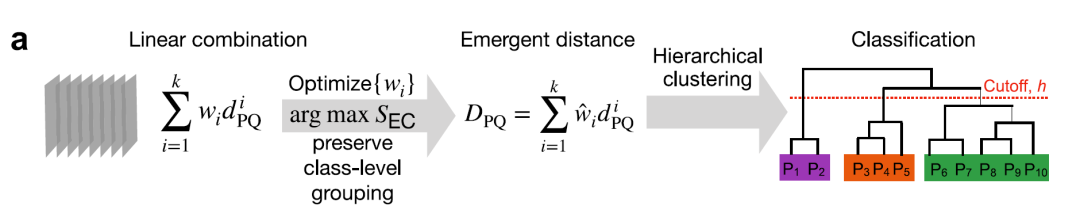
① Distanzmessung in verschiedenen Maßstäben
Die Forscher kodierten paarweise Beziehungen zwischen E3-Ligasen, indem sie 12 verschiedene Distanzen berechneten.Diese Distanzen umfassen mehrere granulare Ebenen: Primärsequenz, Domänenarchitektur, Tertiärstruktur, Funktion, subzelluläre Lokalisation und Zelllinien-/Gewebeexpression.Alle Distanzmetriken werden zum Vergleich und zur Kombination auf das Intervall [0,1] skaliert, wie in der folgenden Abbildung dargestellt:
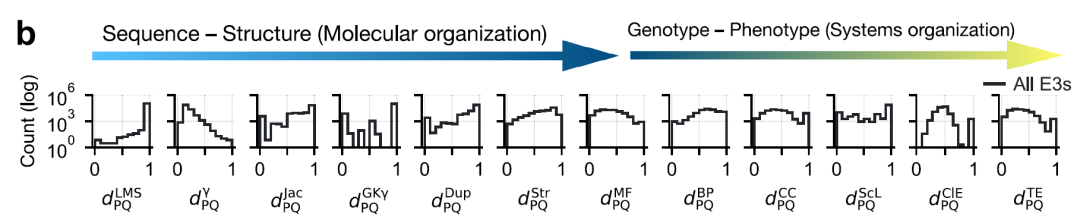
* Sequenzebene: Es wurden die Distanz des lokalen Matching Score (LMS) (ohne paarweises Matching) und die auf der Ausrichtung basierende γ-Distanz verwendet.
* Auf der Ebene der Domänenarchitektur wurden drei Distanzen berechnet: die Jaccard-Distanz, die Goodman-Kruskal-γ-Distanz und die Domänenwiederholungsdistanz.
* 3D-Strukturebene: Verwendung des AlphaFold2-Modells (TM-Score)
* Funktionelle Ebene: Die funktionelle Distanz zwischen Proteinen und P und Q wird anhand der semantischen Ähnlichkeit von GO-Annotationen gemessen, die drei Ontologien umfassen: * Molekulare Funktion (MF), biologischer Prozess (BP) und zelluläre Komponente (CC).
* Subzelluläre Lokalisierungsdistanz
* Koexpressionsdistanz zwischen Geweben und Zelllinien
②Metrische Optimierung, Clustering, Bootstrapping und Klassifizierung
Die vier Hauptdistanzen (γ, Jaccard, Struktur und Molekülfunktion) werden gewichtet und integriert, wobei die Gewichte durch schwach überwachtes Lernen und den Elementzentrumsähnlichkeitsindex (SEC) optimiert werden, wie in der folgenden Abbildung dargestellt, um den optimalen kombinierten Index zu erhalten.
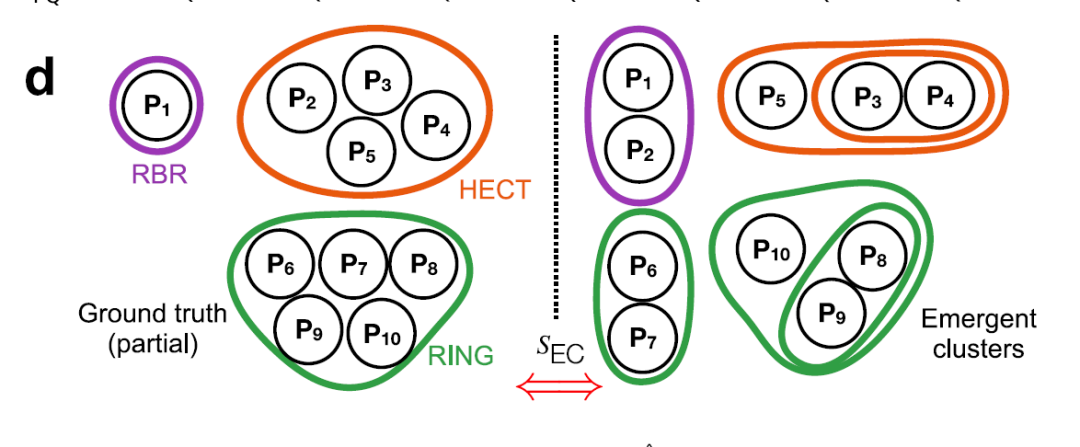
Die hierarchische Clusteranalyse wurde mit der Ward-Methode der minimalen Varianz durchgeführt.Die Unterstützung wird mithilfe der Bootstrapping-Methode berechnet, um das endgültige E3-Dendrogramm zu erstellen. Optimale Cluster werden mit einem Schwellenwert für die Baumsegmentierung von h = 0,25 ermittelt, wodurch die 462 E3-Cluster systematisch in 13 Familien unterteilt werden: 10 RING-Familien, 2 HECT-Familien und 1 RBR-Familie, wie in der Abbildung unten dargestellt.
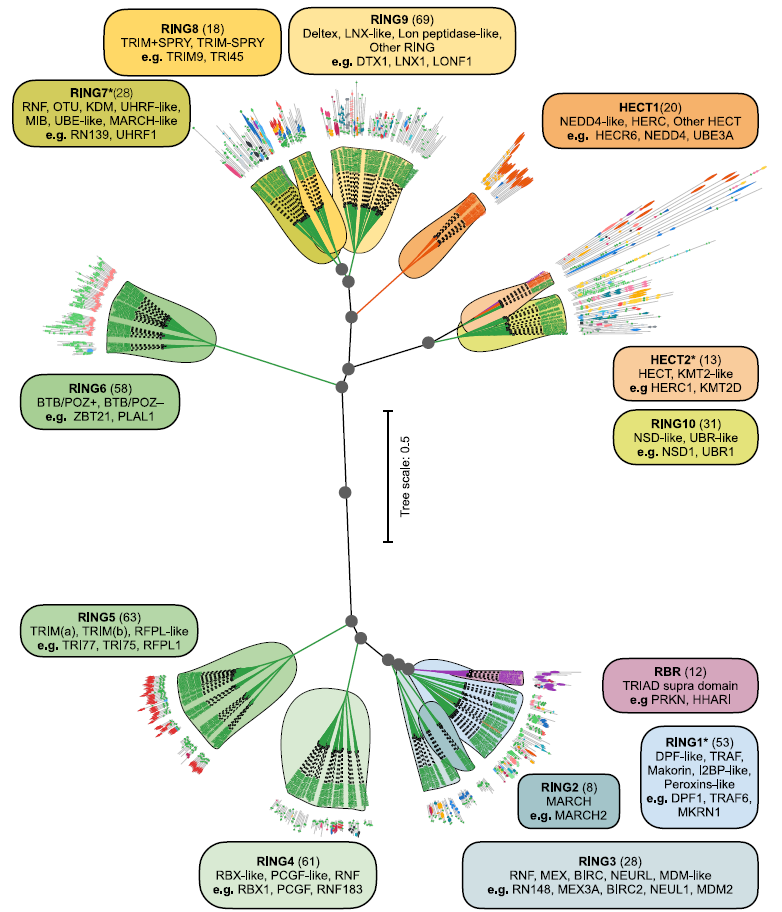
Jede Familie wird einer weiteren manuellen Analyse der Sequenz- und Domänenmerkmale unterzogen, um Unterfamilien und abweichende Proteine zu identifizieren.
③ Wahrscheinlichkeit der Clusterbildung und Bindung kleiner Moleküle
Integrierte 2D-UMAP-Projektion für die Clusterbildung kleiner MoleküleDurch die Kombination lokaler Dichtemaxima wurden zwanzig repräsentative kleine Molekülcluster identifiziert.Die Wahrscheinlichkeit, dass jedes Cluster an das E3-Protein bindet, wurde durch logarithmisch transformierte Propensitäten (LPij) quantifiziert, was als Grundlage für die nachfolgende PROTAC-Entwicklung und das gezielte Design kleiner Moleküle dient.
Es wurde eine detaillierte Bewertung der Integrität des menschlichen E3-Ligase-Genoms vorgelegt.
① Detaillierte Organisation des menschlichen E3-Ligase-Genoms
Um den Herausforderungen durch die unterschiedlichen Strategien und uneinheitlichen Definitionen in bestehenden Studien zur Organisation des E3-Systems zu begegnen, definierte dieses Forschungsteam die katalytischen Komponenten des E3-Systems explizit als Polypeptidsequenzen mit einer oder mehreren katalytischen Domänen. Dieser objektive Standard ermöglicht eine korrekte Annotation und gezielte Analyse des E3-Systems.Letztendlich stellten die Forscher fest, dass 462 Polypeptidsequenzen im gesamten Datensatz mindestens eine katalytische Domäne enthielten.Diese Polypeptide bilden ein fein organisiertes humanes E3-Ligase-Genom, wie in der folgenden Abbildung dargestellt:
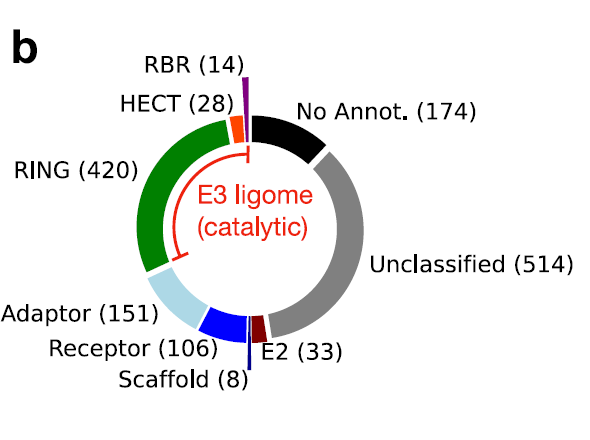
Um die Zuverlässigkeit des Sortierprozesses zu überprüfen, definierten die Forscher für jedes Protein einen Konsenswert, der auf der Häufigkeit seines Vorkommens in Datensätzen aus verschiedenen Quellen basiert.Die Ergebnisse zeigten, dass die HECT- und RBR-Klasse-E3-Ligasen im Datensatz eine hohe Konsistenz aufwiesen (Konsens-Score ≥ 0,6, orange und violette Balken).Die Konsenswerte für die RING-Klasse (grüne Balken) streuen stark, was auf eine Herausforderung bei der Annotation hinweist, wie in der folgenden Abbildung dargestellt:
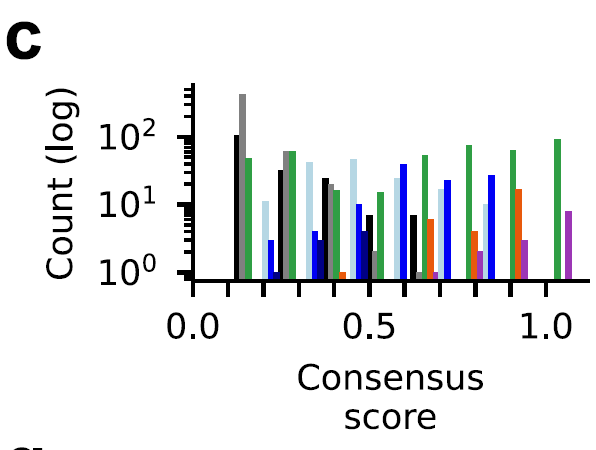
Mit dieser Methode minimierten die Forscher falsch positive und richtig negative Ergebnisse, bezogen hochzuverlässige katalytisch aktive E3-Ligasen mit ein und berücksichtigten Pseudo-E3-Ligasen sowie andere E3-Ligasen, deren katalytische Aktivität nicht verifiziert worden war. Dadurch ermöglichten sie eine detaillierte Beurteilung der Integrität des menschlichen E3-Ligase-Genoms.
② Funktionelle Differenzierung der humanen E3-Ligase
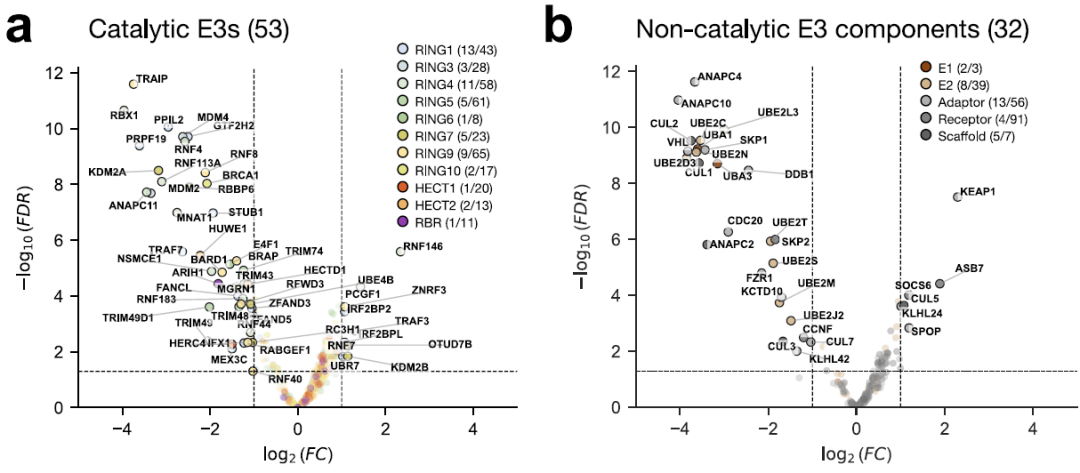
Um die Funktion der menschlichen E3-Ligase zu untersuchen, suchten Forscher nach CRISPR-Cas9-vermittelten Deletionen im UPS-Gen und nutzten die Zellviabilität als primären Phänotyp. Die Ergebnisse zeigten, dass…Insgesamt wurden 53 katalytische und 32 nicht-katalytische E3-Komponenten als entscheidend für die Zelllebensfähigkeit identifiziert.Wie unten gezeigt:
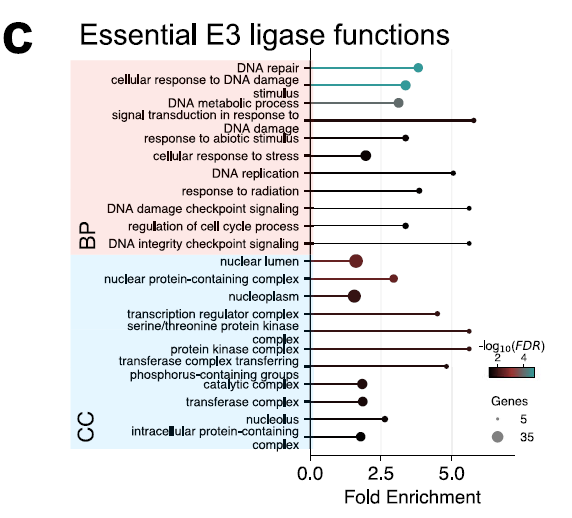
Die GO-Analyse von 53 wichtigen E3-Ubiquitin-Ligasen zeigte eine signifikante Anreicherung in nukleären Komponenten sowie in DNA-Schadens-, Replikations- und Reparaturprozessen (siehe Abbildung unten). Dies deutet auf ihre zentrale Rolle bei der Aufrechterhaltung der Genomstabilität und der nukleären Regulation hin. Diese Ergebnisse identifizieren die für das Zellüberleben essenziellen E3-Ubiquitin-Komponenten.
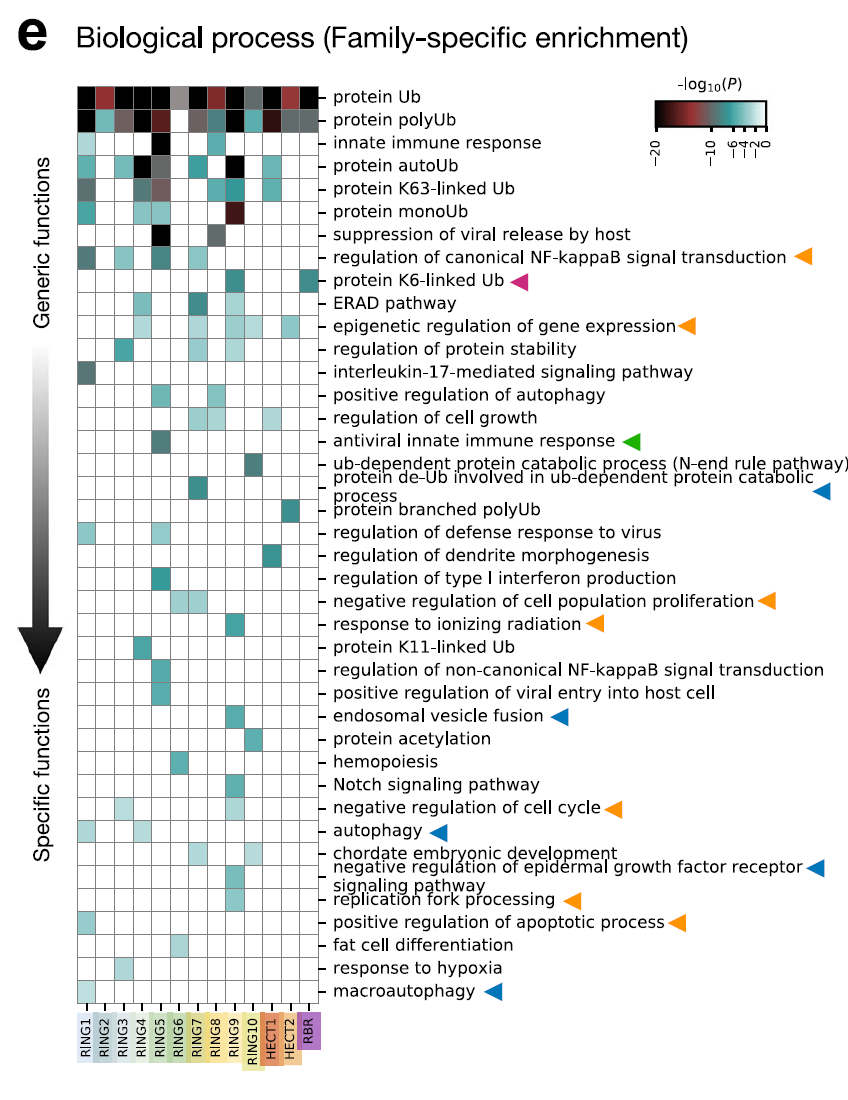
Mithilfe von Metascape wurde eine GO-Anreicherunganalyse an 13 E3-Familien durchgeführt und das Netzwerk mit Cytoscape visualisiert. Die Ergebnisse zeigen, dass…Verschiedene Familien haben unterschiedliche Rollen bei der Substratauswahl, der zellulären Lokalisierung und der katalytischen Funktion.Wie in der Abbildung unten dargestellt. Beispielsweise sind die RBR-Familienmitglieder RNF14, RNF144A und PRKN spezifisch für K6-verknüpftes Ubiquitin. Die K6-verknüpfte Kette kann blockierte RNA-Protein-Quervernetzungen (RNF14), den DNA-erkennenden Adapter STING (RNF144A) zur Aktivierung der Interferon-Signalübertragung und beschädigte Mitochondrien zur Beseitigung (PRKN) markieren.TRIM E3s (RING5) sind in antiviralen angeborenen Immunantworten signifikant angereichert und regulieren die Aktivität von Mustererkennungsrezeptoren in Zellen.Beispielsweise Reaktionen, die durch RIG-1 und MDA5 vermittelt werden.
④ Karte der Arzneimittelwirksamkeit humaner E3-Ligasen
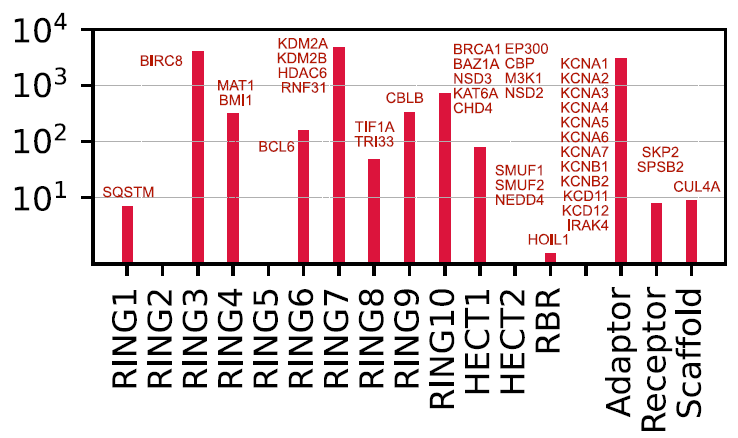
Um potenzielle therapeutische Ansätze basierend auf proximaler Wirkung zu erforschen, ordneten Forscher bestehende E3-Operanden bekannter Proteinabbau-Targeting-Chimären (PROTACs) und E3-Binder verschiedenen E3-Ubiquitin-Ligasen und ihren Familien zu. Derzeit können nur 16 Proteine (9 katalytische E3-Ubiquitin-Ligasen und 7 Adapterproteine) direkt durch bestehende E3-Operanden adressiert werden. Die meisten der entwickelten E3-Operanden zielen auf Adapterproteine (wie VHL und CRBN), während nur sehr wenige direkt katalytische E3-Ubiquitin-Ligasen (wie XIAP, MDM2/4/7 und BIRC2/3/7) adressieren.
Eine Nearest-Neighbor-Analyse mit der humanen E3-Ligase aus dieser Studie ergab fünf stark korrelierte Proteine (BIRC8, RN166/181/141 und UBR2).Wie unten gezeigt:
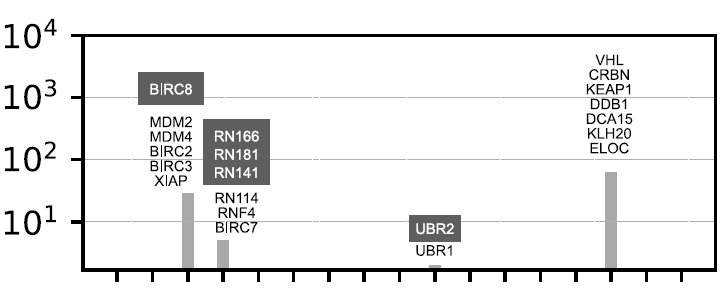
Aufgrund ihrer hohen strukturellen Ähnlichkeit (oftmals homologe Proteine) können bestehende E3-Operanden wiederverwendet werden, um diese Proteine gezielt anzusteuern.Die Kartierung von niedermolekularen E3-Bindern hat Forschern Zugang zu einer potenziellen Reihe von Verbindungen verschafft, die auf weitere 25 E3-Moleküle und 15 nicht-katalytische Komponenten abzielen können.Diese Entdeckung deckt bisher ungenutzte Zielstrukturen auf und bietet einen Weg für die rationale Entwicklung von Leitstrukturen für den E3-Operanden, wie in der folgenden Abbildung dargestellt:
Multiskalige Rahmenwerke bieten leistungsstarke Werkzeuge für die Analyse komplexer biologischer Systeme.
Im Bereich des maschinellen Lernens bezeichnet ein Multi-Scale-Framework eine Modellierungsmethode oder Analysestrategie, die Daten auf verschiedenen Abstraktionsebenen oder Merkmalsskalen verarbeiten kann. Es handelt sich dabei nicht um einen festen Algorithmus, sondern um ein Designkonzept zur Integration lokaler und globaler Informationen sowie grob- und feinkörniger Merkmale, wodurch die Ausdrucksstärke und Generalisierungsfähigkeit des Modells verbessert werden.
Der Wert des multiskaligen Klassifizierungsmodells beschränkt sich nicht auf die systematische Untersuchung der E3-Ligase-Familie; seine Bedeutung liegt vielmehr darin, ein übertragbares und skalierbares Paradigma für die Omics-Integration bereitzustellen. Dieser skalenübergreifende Integrationsansatz ermöglicht die Erweiterung auf andere multimodale Omics-Daten und bietet somit ein universelles Werkzeug für die systematische Analyse komplexer biologischer Systeme.
Die Zelle ist beispielsweise die Grundeinheit des Lebens, und ihre Funktion und ihr Schicksal werden von komplexen molekularen Netzwerken bestimmt. Traditionelle Deep-Learning-Methoden eignen sich zwar gut zur Zelltypidentifizierung anhand von Einzelzell-Transkriptomdaten, es mangelt ihnen jedoch an biologischer Interpretierbarkeit. Am 20. Oktober 2025 stellten Forscher des Nationalen Proteinwissenschaftszentrums Chinas (Peking) und der Tsinghua-Universität den Cell Decoder vor, ein mehrskaliges, interpretierbares Deep-Learning-Framework, das biologisches Vorwissen integriert.Es ermöglicht die hierarchische Charakterisierung und das Schlussfolgern von Genen und Signalwegen auf biologische Prozesse und bietet damit einen neuen Ansatz zur Entschlüsselung von Zelltypen auf Einzelzellebene. Cell Decoder erstellt einen skalenübergreifenden biologischen Wissensgraphen, indem er Proteininteraktionsnetzwerke, Gen-Pfad-Zuordnungen und hierarchische Pfadbeziehungen in eine Graph-Neuronale-Netzwerk-Architektur einbettet.Das Forschungsteam evaluierte Cell Decoder systematisch anhand von menschlichen und Mausproben aus sieben öffentlich zugänglichen Einzelzelldatensätzen im Vergleich zu neun etablierten Methoden. Die Ergebnisse zeigten, dass Cell Decoder sowohl hinsichtlich der Vorhersagegenauigkeit (0,87) als auch des Macro F1-Werts (0,81) den ersten Platz belegte und selbst unter komplexen Bedingungen wie Rauschen, unausgewogenem Zelltyp und Verschiebungen in der Verteilung zwischen verschiedenen Datensätzen eine stabile Leistung beibehielt.
Titel des Papiers:Zelldecoder: Entschlüsselung der Zellidentität mit multiskaligem, erklärbarem Deep Learning
Papieradresse:
https://link.springer.com/article/10.1186/s13059-025-03832-y
Langfristig lässt sich das Multi-Skalen-Framework mit räumlichen Proteomikdaten, Wirkstoffbibliotheken niedermolekularer Substanzen und chemischen Rauminformationen integrieren und so Datenbarrieren zwischen biologischer Grundlagenforschung, Krankheitsmechanismusanalyse und translationalen Anwendungen überwinden. Mit der kontinuierlichen Zunahme von Multi-Omics-Daten wird diesem Framework voraussichtlich eine immer wichtigere Rolle in der lebenswissenschaftlichen Forschung und biomedizinischen Innovation zukommt.
Quellen:
1.https://www.nature.com/articles/s41467-025-67450-9
2.https://blog.csdn.net/qazplm12_3/article/details/153948711
3.https://link.springer.com/article/10.1186/s13059-025-03832-y