Command Palette
Search for a command to run...
コーネル大学は、高伝導性リチウムイオン電解質の化学メカニズムを解読するための革新的な AI フレームワークを提案し、% で 80% を超える予測成功率を達成しました。

新エネルギー電池市場の急速な拡大、特にリチウムイオン電池、固体電池、高エネルギー密度電池の広範な応用に伴い、電解質性能の最適化は、電池の安全性、効率、寿命を決定する重要な要素となっています。
塩溶媒化学は、ほとんどのリチウムイオン電池システムにおける電解質挙動の基盤であり、イオン伝導性、粘度、化学的安定性といった重要な特性を決定づけます。しかしながら、その合理的な設計は、無数の組み合わせと非線形な構造-性能相関関係を包含する広大な化学空間によって制約されています。実験データがまばらで不均一に分布していることは、この問題をさらに悪化させ、モデルの一般化能力を阻害します。近年、AIによる自律的な電解質発見は一定の進歩を遂げていますが、しかし、既存の研究では、大規模な電解質配合によってカバーされる広大な化学空間を探索しながら、固有の解釈可能性を維持できる統一された計算パラダイムが依然として明らかに欠けています。
この文脈では、コーネル大学の研究チームは、塩溶媒化学をモデル化および解釈するための堅牢で解釈可能、かつデータ効率の高いフレームワーク SCAN を開発しました。このフレームワークは、ロングテールデータを効果的に処理し、塩溶媒配合の全範囲を網羅します。研究者らはSCANを非水電解質(NAE)システムに適用し、導電率予測におけるベースライン誤差を0.372 mS·cm⁻¹に抑え、ベースラインモデルと比較して予測誤差を65.31 TP³T削減しました。
さらに重要なことは、大規模な検証により、このモデルは、上位候補システムに対して 81.08% の予測成功率を達成しました。SCAN は予測機能に加えて、勾配分離、記号回帰、量子化学計算を導入して化学メカニズムを明らかにし、分子の柔軟性とイオン溶媒相互作用が伝導性に与える影響を明らかにします。
「塩-溶媒化学のための動的ルーティングガイドによる解釈可能なフレームワーク」と題された関連研究成果が Nature Computational Science に掲載されました。
研究のハイライト:
* SCAN は、高性能 NAE 塩溶媒化学研究における重要なギャップを埋めます。
原子中心のポテンシャルエネルギー面モデルにヒントを得て、記述子中心の専用表現と注目メカニズムを備えたマルチ特徴ネットワーク (MFNet) を開発しました。
* 革新的な動的ルーティング戦略が MFNet に導入され、モデルは元のデータ分布を変更することなく、広範囲にわたってイオン伝導率を正確に予測できるようになりました。

用紙のアドレス:
https://www.nature.com/articles/s43588-026-00955-5
弊社の公式 WeChat アカウントをフォローし、バックグラウンドで「SCAN」と返信すると、完全な PDF を入手できます。
データ次元は広範囲にカバーされている
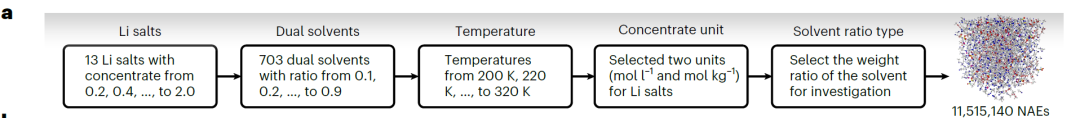
高精度のSCANモデルを訓練するために、研究チームは、13 種類のリチウム塩と 38 種類の有機溶媒 (下図参照) を含む、合計 13,302 件の完全なデータ エントリを含む CALiSol データセットを構築しました。
データの次元は広範囲にわたり、各データ ポイントには次のものが含まれます。
* イオン伝導率 k: 0~38.1 mS/cm
* 温度 T: 194.15~477.42 K
* 塩分濃度 c: 0~4 mol/L または mol/kg
※リチウム塩の種類:LiPF₆、LiTFSI、LiFSI、LiBOBなど、合計13種類。
* 溶剤の種類: エチレンカーボネート (EC)、メチルエチルカーボネート (EMC)、プロピオニトリル (AN) など。
* 混合戦略SRT: モル比、体積比、質量比
すべてのリチウム塩および溶媒の分子情報はSMILES配列を用いて三次元分子座標に変換され、B3LYP/6-31G理論レベルで幾何最適化が行われ、正確で信頼性の高い分子構造と電子特性が確保されました。この方法により、データセットは、イオン伝導率の値だけでなく、各システムの分子特性、構造情報、溶媒和環境も提供します。データの整合性と科学的な解釈可能性のバランスを取りながら、AI モデルに豊富な入力を提供します。
データセットの構築中、研究チームはロングテールデータ (LTD) の問題に特に注意を払いました。つまり、高伝導率のシステムの数は限られている一方で、ほとんどのシステムの伝導率は低く、9,115 個の NAE (約 68.5%) で k < 5 mS·cm⁻¹、67 個のみで k > 20 mS·cm⁻¹ (約 0.5%) でした。
SCAN: MFNetと動的ルーティングメカニズムを採用
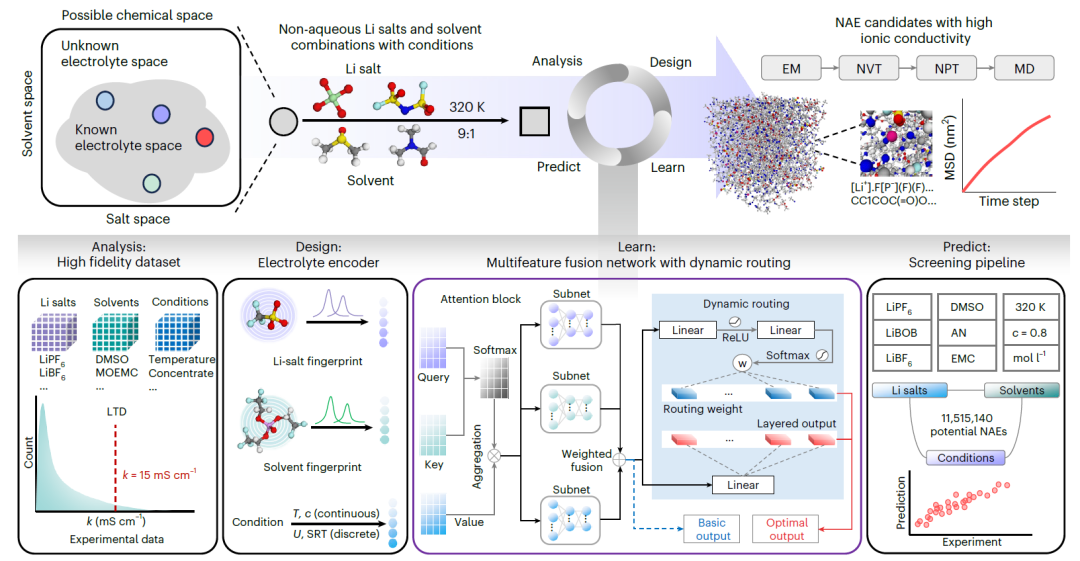
SCANは、動的ルーティングを基盤とするNAEエンジニアリングプラットフォームです。次の図は、その完全なワークフローを示しています。
分析
リチウム塩、溶媒、実験条件の様々な組み合わせを網羅した高忠実度の実験データセットを構築しました。各変数について統計解析を実施し、k値のロングテール分布の特徴付けを行いました。
デザイン
化学情報を持つ分子指紋は、リチウム塩と溶媒それぞれに対して設計され、条件付きコーディングと組み合わされて、塩、溶媒、条件の組み合わせを正確に特徴付けます。
学ぶ
NAEのk値を予測するために、カスタムマルチフィーチャーフュージョンネットワーク(MFNet)が開発されました。塩、溶媒、条件の表現をそれぞれ処理するために、クエリ、キー、値のデータを埋め込むことで独立したアテンションモジュールを構築し、それらを全結合ニューラルネットワークを用いて融合しました。さらに、モデルの性能と堅牢性を向上させるために、動的ルーティング戦略が導入されました。
予測する
1,000万通り以上の塩と溶媒の組み合わせから潜在的なNAEを同定するためのハイスループットスクリーニングパイプラインを構築しました。高い予測k値を持つ候補系は、エネルギー最小化(EM)、カノニカルアンサンブル(NVT)、等温等圧アンサンブル(NPT)、MD生成シミュレーション、平均二乗変位(MSD)解析、およびラジアル分布関数(RDF)計算を含む分子動力学(MD)シミュレーションを用いてさらに検証されました。
MFNet と動的ルーティング メカニズムはフレームワーク全体の中核であり、具体的なメカニズムは次のとおりです。
MFNet: マルチチャネル自己注意ネットワーク
原子中心ポテンシャル面モデルにヒントを得た MFNet (分子特徴ネットワーク) フレームワークは、ネットワークを 3 つの独立したサブネットに分割し、それぞれが異なる機能を処理します。
* リチウム塩サブネット: CALiSol データセットでは、各 NAE には 1 種類のリチウム塩のみが含まれているため、その記述子ベクトルはリチウム塩サブネットに直接入力されます。
* 溶媒サブネット: 一部のデータ ポイントには複数の溶媒が含まれるため (例: PC と AN が 0.9:0.1 の比率で混合)、これらの溶媒記述子の平均値が入力として計算され、全体的な溶媒環境が反映されます。
* 条件付きサブネット: 温度や濃度などの実験条件を処理します。
自己注意モジュールに続いて、2つの全結合隠れ層を用いて入力の漸進的投影と非線形変換を行い、高次元特徴量を生成することで、後続処理の表現力を高めます。層間にはReLU非線形活性化関数を用いることで、モデルの表現力をさらに向上させます。
最終的なネットワークアーキテクチャは、リチウム塩サブネット(14–16–16)、溶媒サブネット(14–16–16)、条件付きサブネット(6–16–16)です。固有次元(特徴次元<128)を持つ潜在的な出力は、重み付き融合モジュールに入力され、入力特徴のグローバルな依存関係情報を保持しながら重み付き出力を計算します。リチウム塩、溶媒、条件付き特徴のエンコードは3つのサブネットによって独立して処理されるため、シングルヘッドセルフアテンション(SHA)を使用して小規模な特徴データ(それぞれ14、14、6次元)を軽量に処理でき、埋め込み次元の低いタスクに適しています。
動的ルーティングメカニズム:ロングテールデータの問題を解決する
ロングテール分布では、データサンプルは主に「ヘッド領域」に集中し、「テール領域」のサンプルは不足しています。しかし、従来のモデルはヘッドサンプルに過剰適合し、テール領域を無視することが多く、結果として、重要だが不足している化学空間の探索が不十分になります。この問題に対処するため、本研究ではMFNetに動的ルーティング戦略を導入します。すべてのサンプルを平等に扱う標準的なアーキテクチャとは異なり、動的ルーティングは、レイヤー入力に基づいて異なるレイヤーに異なる表現機能を適応的に割り当てるためのソフト ゲーティング メカニズムを学習します。以下に示すように:

このメカニズムにより、希少サンプルは異なるルーティングパスをアクティブ化し、条件付き計算を容易にすることで、低頻度カテゴリの一般化能力を向上させることができます。その主な特徴は以下の2つです。
* 入力依存ルーティング重み: つまり、元の損失関数またはデータ分布を変更せずに各サンプルの特徴サブスペースを選択します。
* カテゴリ適応型機能分離: これには、主要なカテゴリと末尾のカテゴリ間の違いを明示的にモデル化することが含まれ、動的ルーティングは単純な静的損失再形成よりも柔軟で解釈可能な LTD ソリューションを提供することを示します。
解釈可能性:GBAと記号回帰
SCAN は、イオン伝導率を正確に予測しながら化学的解釈可能性を提供します。
* GBA(勾配ベース属性):SCANモデルは3並列アテンションニューラルネットワークフレームワークを採用しているため、その意思決定プロセスの可視化はツリーモデルと比較して複雑です。kに影響を与える主要な化学的因子を特定するために、GBA法を用いて特徴量の重要度を評価しました。これは、各入力特徴量のモデル出力への勾配寄与を計算し、最も重要なリチウム塩、溶媒、および条件的特徴量を特定するものです。
* シンボリック回帰:主要な Li 塩、溶媒、条件情報と kkk との間の解釈可能な機能的関係を発見するために、PySRRegressor に基づく記号回帰法が使用されました。
SCAN モデルは、すべてのベースライン モデルを一貫して上回ります。
MFNet と動的ルーティングを使用した SCAN フレームワークのパフォーマンスを総合的に評価するために、研究者は MFNet に基づく 4 つのベースライン モデルを構築しました。いずれも動的ルーティングを含んでいない: (1) 基本MFNetモデル: B1 (未処理記述子) と B2(最大値でスケールされた記述子);(2)少数クラスオーバーサンプリング技術SMOTEを組み合わせたMFNetモデル:S1–S18(3)K近傍アンダーサンプリング法(KUTE)を組み合わせたMFNetモデル:U1–U9(4)SMOTEとKUTEを組み合わせたMFNetモデル:SU1–SU9 。
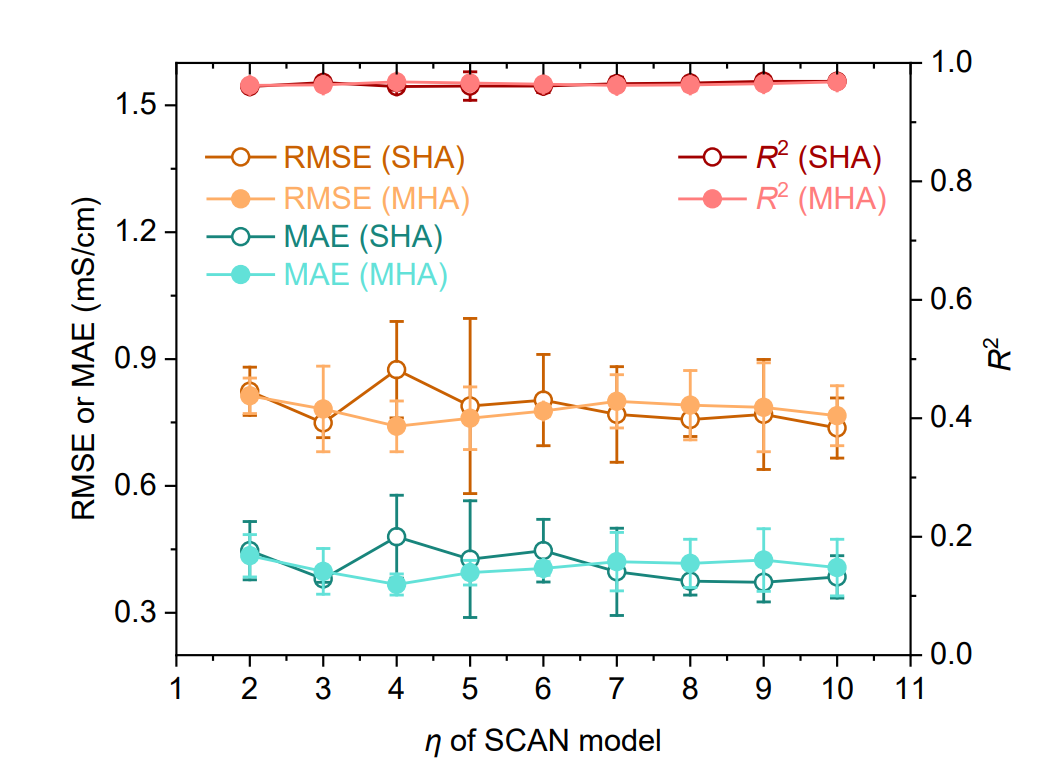
同時に、下図に示すように、Transformerベースのモデル、グラフニューラルネットワーク(GNN)、サポートベクター回帰(SVR)、およびエクストリーム勾配ブースティング(XGBoost)をベースライン比較として使用しました。さらに、シングルヘッドアテンション(SHA)またはマルチヘッドアテンション(MHA)構造と、異なる数の動的ルーティング層(η = 2~10)を採用したSCANモデルを実装し、モデルの堅牢性を検証しました。
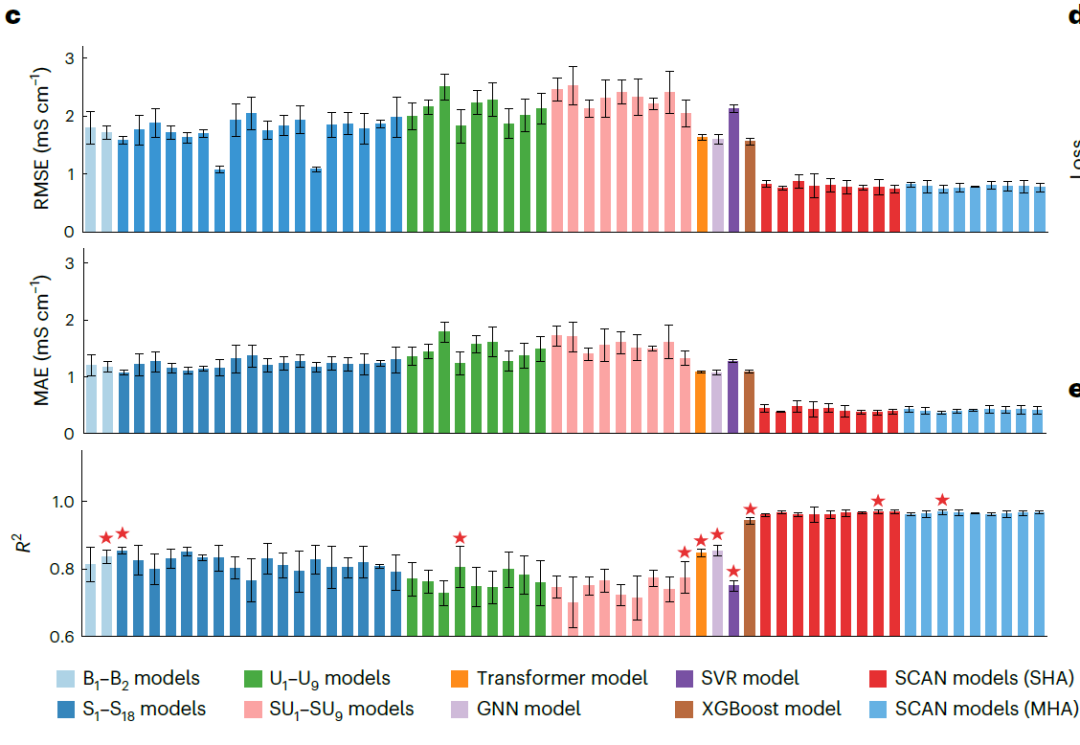
NAEパフォーマンス評価
下図に示す結果は、SHA(η = 9)を用いたSCANモデルが全てのベースラインモデルを一貫して上回り、予測誤差(RMSE 0.769 mS·cm⁻¹、MAE 0.372 mS·cm⁻¹)が大幅に低下し、R²(0.969)も高いことを示しています。注目すべき点は…MAE における最高パフォーマンスのベースライン GNN (1.072 mS·cm⁻¹) と比較すると、誤差は 65.31 TP3T 減少しました。MHA (η = 4) を使用した SCAN パフォーマンスは SHA バージョンに匹敵し、MFNet が動的ルーティングとの統合において優れた堅牢性を備えていることを示しています。
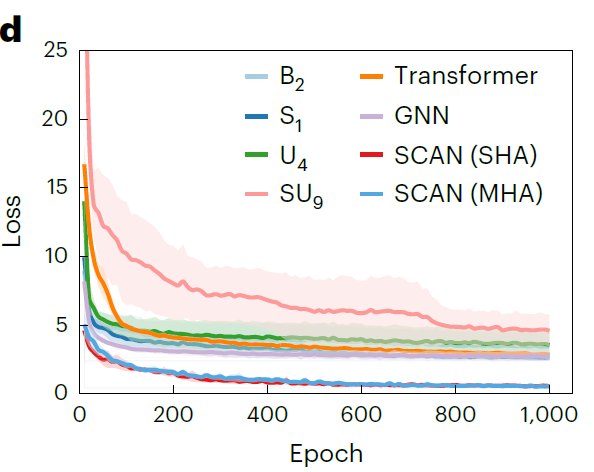
また、研究者らは、1,000 回のトレーニング エポックにわたって SCAN と代表的なベースライン モデルの検証損失曲線をプロットしました。下の図に示すように、トレーニングの開始時から、SCAN の予測誤差はベースライン モデルよりも大幅に低く、初期損失はわずか 4.59 mS·cm⁻¹ であり、検証損失は減少し続け、さらに低いレベルに収束しており、より強力な安定性と一般化能力を備えていることがわかります。
高スループットNAEスクリーニング機能
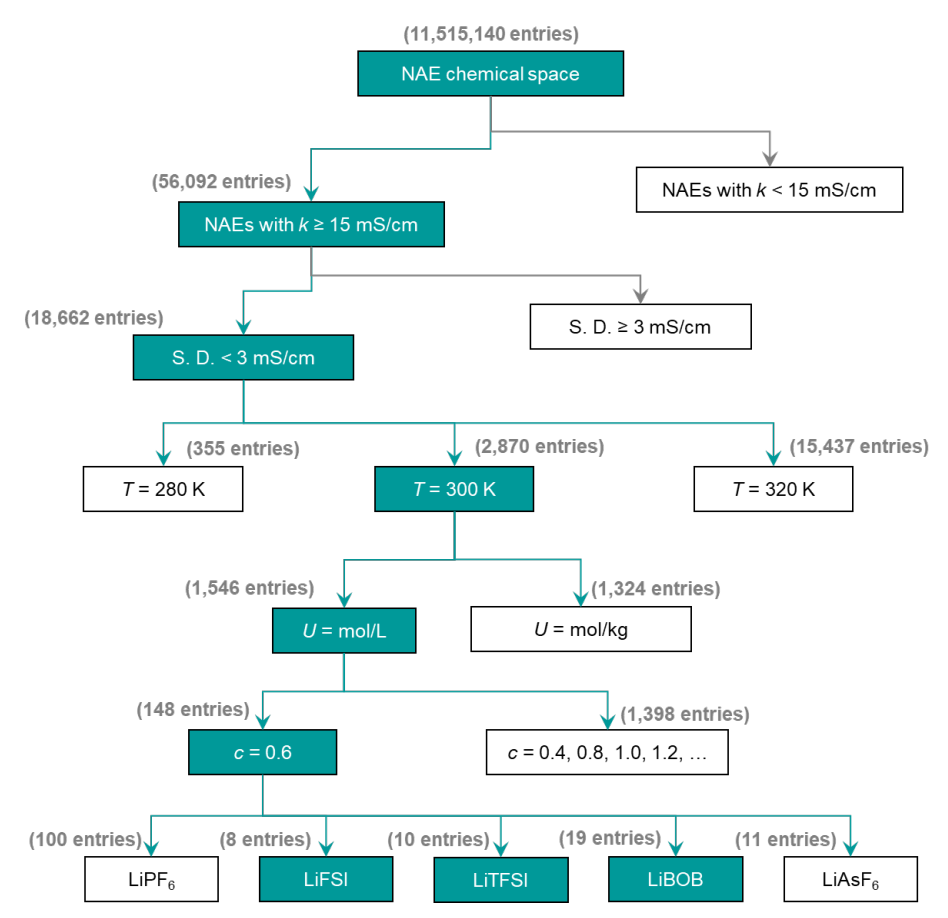
高精度スキャンは、NAEの拡張された化学空間を効率的に探索することを可能にします。この目的のために、研究者らは高kNAEを特定するために設計されたハイスループットスクリーニングプロセス(下図参照)を開発しました。これに基づいて、11,515,140 の潜在的な二重溶媒 NAE が生成されました。
SCANの予測力を活用することで、k ≥ 15 mS·cm⁻¹のNAE 56,092件を迅速に特定し、そのうち18,662件は予測不確実性が低い(<3 mS·cm⁻¹)ことが分かりました。このプロセスにより、分子動力学シミュレーションと実験の計算負荷とコストが大幅に軽減されます。
候補非水電解質(NAE)の検証
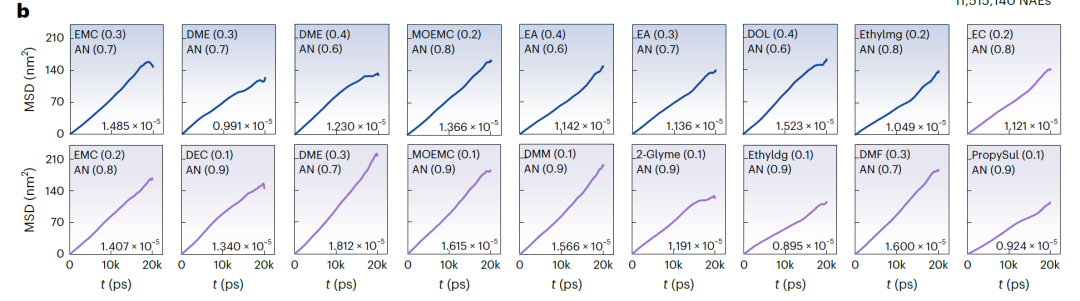
研究者らは、予測を厳密に検証するために、分子動力学(MD)シミュレーションを実施し、k値を算出しました。温度(T)、濃度(c)、および実際の条件に基づいて、MD検証範囲をLiFSI、LiTFSI、LiBOBに基づくNAE候補系に絞り込み、最終的に下図に示すように、詳細な研究のために37の有望な系を選択しました。
各システムの平均計算時間は約10時間(36,355.12秒)で、10⁷個の候補システムをシミュレーションするのにかかる推定コストは約10⁸GPU時間です。これは、理想的な高性能コンピューティング環境下であっても、総当たり方式による選択の実際的な能力をはるかに超えています。これに対し、適切にトレーニングされた SCAN モデルは、候補システムごとに記述子の計算から最終予測までのプロセス全体を 5 秒未満で完了できるため、計算コストを 7,200 倍以上削減できます。これにより、スケーラビリティと効率性が大幅に向上します。これは、NAE探索におけるSCANフレームワークの必要性を浮き彫りにしています。SCANフレームワークの代替モデルは、極めて低い計算コストで高性能な候補を迅速に優先順位付けできます。
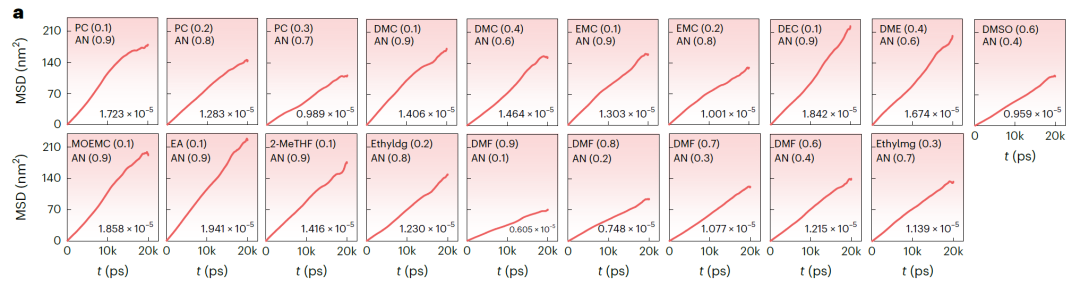
下の図 b (LiFSI および LiTFSI に基づく NAE)と図 a (LiBOB に基づく NAE)は、MD シミュレーションから得られた時間の経過に伴う平均二乗変位(MSD)と対応する拡散係数を示しています。すべてのシステムの MSD は安定した線形増加を示しており、シミュレーションが安定して収束していることを示しています。SCAN によって予測された k 値は MD シミュレーション結果と非常に一致しており、平均偏差はわずか 3.198 mS·cm⁻¹、最大偏差は 7.342 mS·cm⁻¹ です。検証システムでは、k > 15 mS·cm⁻¹ の 25 システムで成功率は 67.571TP⁻¹ でした。予測された k > 14 mS·cm⁻¹ のシステムでは、検証成功率は 81.081TP⁻¹ に増加しました。
人工知能は、バッテリーの研究開発の基本的なパラダイムを再構築しています。
電池は、民生用電子機器、電気自動車、系統電力貯蔵システムといった主要用途の中核部品として、世界的な持続可能なエネルギー転換を牽引しています。エネルギー密度と電力密度の向上、サイクル寿命の延長、安全性の向上、そして製造コストの削減は、電池の研究開発の中核目標です。これらの目標達成の鍵は、電池内部の電気化学メカニズム、具体的には電気化学界面反応と安定化メカニズム、電子とイオンの結合と輸送、そして次世代電極材料のエネルギー貯蔵メカニズムを深く理解することです。
技術進化のより広い視点から見ると、SCANフレームワークに代表される「データ駆動型+解釈可能なモデリング」パラダイムは、次世代バッテリー研究開発システムの重要な構成要素になりつつあります。過去数十年にわたり、バッテリー材料システムのイノベーションは主に経験主導型で試行錯誤的な実験に依存しており、開発サイクルの長期化と高コスト化を招いていました。しかし、機械学習とハイスループットコンピューティングの統合により、バッテリー研究開発はますます効率化しています。
例えば、ByteDance のシードチームの Wen Yan 氏と Sheng Gong 氏らは、前向き予測モデルと逆生成方法を統合した電解質配合設計の統一フレームワークを開発しました。研究者らは、特性タグ付きの単一分子(24万種類以上)および分子混合物(1万種類以上)に関する文献データを広範囲に収集し、電解質設計空間を広くカバーしました。さらに、分子動力学シミュレーションから得られた10万種類以上の分子混合物データを組み込むことで、導電性だけでなく、リチウム金属電池の界面安定性に関連する融解構造も予測できる高精度な機械学習モデルを構築しました。
論文タイトル: 液体電解質配合のための統合予測・生成ソリューション
論文リンク:
https://www.nature.com/articles/s42256-025-01173-w
総じて、人工知能はバッテリー研究開発の根底にあるパラダイムを再構築しています。モデルと実験システムの深い統合により、バッテリー材料の発見速度とイノベーション密度は新たな加速サイクルに入ると予想されます。
参考文献:
https://www.nature.com/articles/s43588-026-00955-5
https://phys.org/news/2026-02-ai-framework-reveals-chemistry-high.html
https://static-content.springer.com/esm/art%3A10.1038%2Fs43588-026-00955-5/MediaObjects/43588_2026_955_MOESM1_ESM.pdf
https://www.eet-china.com/mp/a471613.html








