Command Palette
Search for a command to run...
AIを活用した量子精密化:カーネギーメロン大学などが、量子力学的制約を用いてタンパク質の全原子モデルを精密化する初の手法であるAQuaRefを提案した。
生命過程の分子メカニズムを理解するためには、まず生体高分子の三次元構造を把握する必要がある。原子レベルの構造を決定することは、構造生物学の中核的な課題であり、タンパク質の機能の理解、遺伝子制御機構の解明、標的薬の開発のための重要な基盤となる。タンパク質触媒反応、核酸による遺伝情報の伝達、抗体による抗原認識など、これらの重要な生物学的プロセスはすべて、説明のために精密な構造モデルに依存している。
現在、生体高分子の構造解析には、クライオ電子顕微鏡法とX線結晶構造解析が主要な実験手法として用いられており、高分解能の構造データが大量に蓄積されている。近年では、AlphaFoldやRoseTTAFoldに代表される計算予測手法も著しい進歩を遂げ、構造モデリングのための効率的なツールを提供している。しかしながら、未知の構造タイプの発見や複雑な相互作用の解明においては、実験解析は依然として不可欠な役割を果たしている。実験的な構造解析プロセスにおいて、原子モデルの精密化は最終段階に近い重要なステップである。その目的は、立体化学の法則に適合し、実験データにできる限り近い分子モデルを構築することである。CCP4やPhenixなどの現在の主流の精密化ソフトウェアは、主に標準データベースの立体化学的制約に依存して、妥当な結合長と結合角を維持し、原子間の衝突を低減している。
しかしながら、このような制約システムには依然として重大な限界がある。それらは主に共有結合構造を対象としており、水素結合やπパッキングなどの重要な非共有結合相互作用の体系的な記述が欠けている。低解像度では、これがモデルが真の化学状態から逸脱する原因となる可能性がある。構造中に新しいリガンドや特異な結合が現れた場合、精密化のために手動でのパラメータ定義が必要となる。さらに、局所的な化学環境によって引き起こされる妥当な幾何学的偏差が、制約システムによって異常と誤って解釈され、強制的に修正される可能性がある。理論的には、量子力学は分子間相互作用をより正確に記述できるが、生体高分子は通常、数千個、あるいは数万個もの原子を含んでいるため、完全な量子コンピューティングは非常に高価になる。したがって、既存の研究のほとんどは、リガンド結合部位などの局所的な領域に限定されている。
この問題に対処するため、カーネギーメロン大学、ポーランドのヴロツワフ大学、フロリダ大学などの大学からなる共同研究チームが、AIを活用した量子精密化手法であるAQuaRefを提案する。この手法は、原子ポテンシャル関数の機械学習であるAIMNet2に基づいており、構造精密化タスク向けにカスタマイズされています。古典的な力場の計算効率に近づきながら、量子力学的計算の結果をより正確に近似することができ、生体高分子の全原子量子精密化のための新たな技術的道筋を提供します。
関連する研究成果は、「AQuaRef:機械学習によるタンパク質構造の量子精密化」と題され、Nature Communications誌に掲載された。
研究のハイライト:
* AQuaRefは、AIMNet2機械学習ポテンシャル関数に基づいて、タンパク質原子モデルの量子精密化を初めて実現しました。
* 61種類の低解像度X線およびクライオ電子顕微鏡モデルのテストにおいて、AQuaRefは57種類のモデルを上回る性能を示しました。
* DJ-1およびYajLタンパク質における短い水素結合の場合、AQuaRefは人間の介入なしに、実験的証拠と一致するプロトン位置を決定することができます。

用紙のアドレス:https://www.nature.com/articles/s41467-025-64313-1
弊社の公式WeChatアカウントをフォローし、バックグラウンドで「AQuaRef」と返信すると、PDF全文を入手できます。
ペプチド機械学習における潜在関数を訓練するための、100万個のサンプルからなるデータセット。
本研究は、機械学習を用いてペプチドシステムの潜在的機能のパラメータ化モデルを構築することを目的とする。したがって、データセットの設計においては、化学組成、立体配座空間、分子間相互作用という3つの側面を体系的に網羅する必要がある。
化学的な観点から言えば、研究者らは、20種類の標準アミノ酸、11種類のプロトン化状態、3種類のN末端修飾、および4種類のC末端修飾を網羅した、SMILES文字列形式の小さなペプチドデータベースを構築した。この基礎の上に、すべての単一ペプチドとジペプチドが列挙され、トリペプチドとテトラペプチドのサブセットがランダムに選択された。さらに、ジスルフィド結合で連結されたペプチドとそのセレン化アナログが生成された。立体配座空間を完全に網羅するために、研究者らはOpenEye Omegaソフトウェアを使用して、キラル中心に制限を課すことなく、ねじれ角の集中的なサンプリングを行い、D型、L型、および混合立体化学ペプチド系にモデルを適用できるようにした。
2~4個のペプチドからなる複合体を構築し、分子間相互作用をシミュレートするために、それらの空間配向をランダムに調整した。データ漏洩の可能性を避けるため、データ生成プロセス全体を通して、天然配列や実験構造は参照しなかった。計算規模を制御するため、すべてのペプチドとその複合体における原子の総数(水素原子を含む)は120個に制限した。
初期構造を取得した後研究者たちはまず、GFN-FF力場を用いて分子動力学シミュレーションを行い、非平衡構造をサンプリングした。これは、デカルト座標で拘束することで全体の構成を初期入力に近い状態に維持しつつ、ねじれ角と分子間自由度を解放する。
続いて、委員会方式のクエリによるアクティブラーニング戦略が導入されました。まず、50万個の初期サンプルがランダムに選択され、4つのモデルからなるアンサンブルシステムがトレーニングされました。次に、4回の反復処理が実行されました。各反復処理では、エネルギーと原子間力のモデル予測の不確実性に基づいてサンプルが選択され、これらの不確実性の高い構造がDFT計算後にトレーニングセットに追加されました。最後の反復処理では、不確実性誘導型最適化がさらに導入され、予測の不確実性は高いがエネルギーが低い境界構造が優先されました。このプロセスを経て、最終的に平均約42個の原子を含む約100万個のサンプルからなるトレーニングセットが得られました。
研究者らは、理論的に生成されたデータに加えて、RCSBおよびEMDBデータベースから実験構造をスクリーニングし、モデルの妥当性を検証した。スクリーニング基準には、タンパク質のみを含む単一コンフォメーションモデル、1,000~10,000個の非水素原子、2.5~4Åの分解能、MolProbityの競合スコアが50未満、結合長および結合角の偏差が標準値の4倍を超えないことなどが含まれる。
AQuaRef:高分子システムのためのAI駆動型量子精密化アプローチ
AQuaRefはまず、入力された原子モデルの整合性チェックを実行します。構造中に欠落している原子については、プログラムが自動的に補完を試みます。ただし、この処理によって、特に元のモデルに水素原子が含まれていない場合、新たな立体障害が生じる可能性があります。欠落している原子が主鎖原子などの重要な構造である場合、量子精密化を続行することはできません。重大な立体障害や深刻な幾何学的異常が検出された場合は、まず標準的な立体化学的制約を用いた迅速な幾何学的正則化を実行し、原子位置の最小限の調整で問題を解消します。
結晶学的データの場合、精密化には単位格子の対称性と周期的な相互作用も考慮に入れる必要がある。具体的には、このプログラムは空間群対称演算子に基づいてモデルをスーパーセルに展開し、その後、メインコピー原子からの距離が設定された範囲内にある対称コピーのみを残して、スーパーセルを切り詰めます。このプロセスは、クライオ電子顕微鏡構造では通常不要です。
原子の補完とモデルの拡張が完了すると、システムはQ|Rソフトウェアパッケージの標準的な精密化プロセスに入ります。AQuaRefのコアアーキテクチャは基本的に基本のAIMNet2モデルと同じですが、構造精密化タスクのためにいくつかの重要な調整が加えられています。
まず、このモデルは長距離クーロン相互作用や分散相互作用を明示的に計算するのではなく、DFT-D4の全エネルギーを再現するように直接学習されています。これは、CPCM暗黙溶媒モデルでは、原子電荷のみを用いてクーロン相互作用を正確に推定することが困難であり、また、長距離相互作用が分極可能な連続媒体によって大きく遮蔽されるためである。さらに、カットオフ半径が5Åを超える長距離分散項は、精密化プロセスにおける主要な原子間力にほとんど寄与しないため、精度に影響を与えることなく無視できる。
第二に、このモデルはGFN1-XTBから明示的な短距離指数関数的反発項を導入しており、空間的な立体障害の衝突がある構造を扱う際に、より優れた安定性をもたらします。モデルの学習は、B97M-D4/def2-QZVP法で計算されたエネルギー、原子間力、およびハーシュフィールド部分原子電荷を用いて行われた。学習は、ランダムな重み初期化、バッチサイズ256、合計150万ステップの学習で開始された。その他のハイパーパラメータはすべて、元のAIMNet2の設定から引き継がれた。
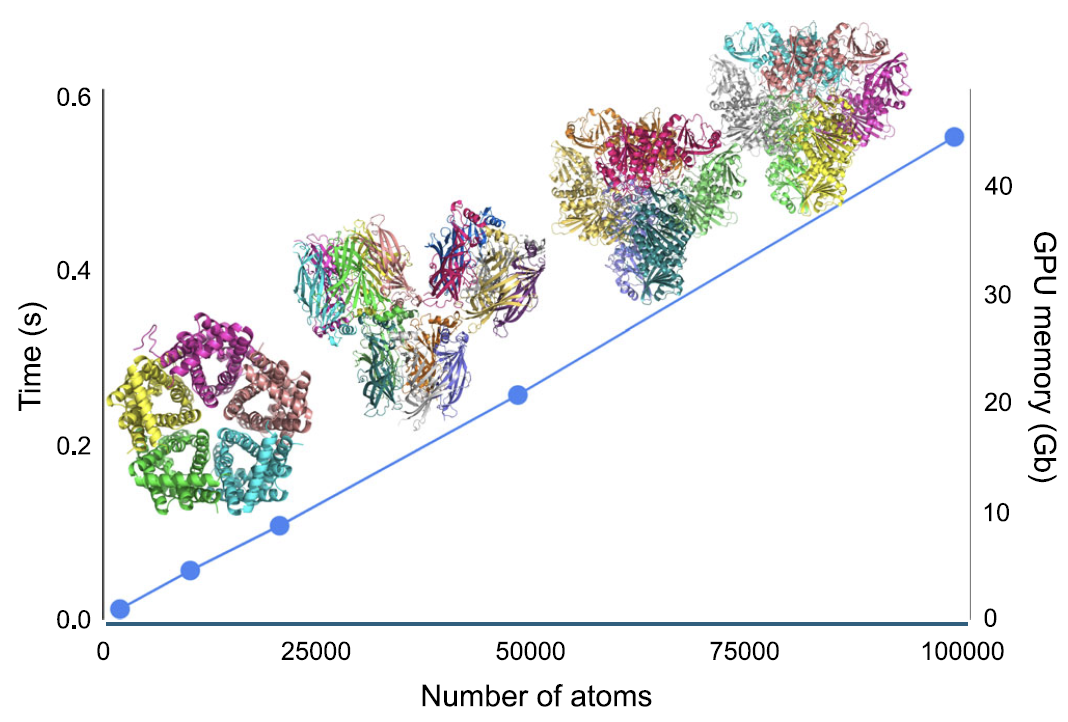
計算効率に関しては、下図に示すように…AIMNet2フレームワークでは、エネルギーと原子間力の計算時間、およびGPUメモリのピーク使用量はすべて、システム内の原子数に対して線形(O(N))に増加します。約10万個の原子を含むタンパク質系の場合、一点エネルギーおよび力の計算は約0.5秒しかかかりません。80GBのビデオメモリを搭載した単一のNVIDIA H100 GPUでは、最大約18万個の原子を含むモデルを処理できます。
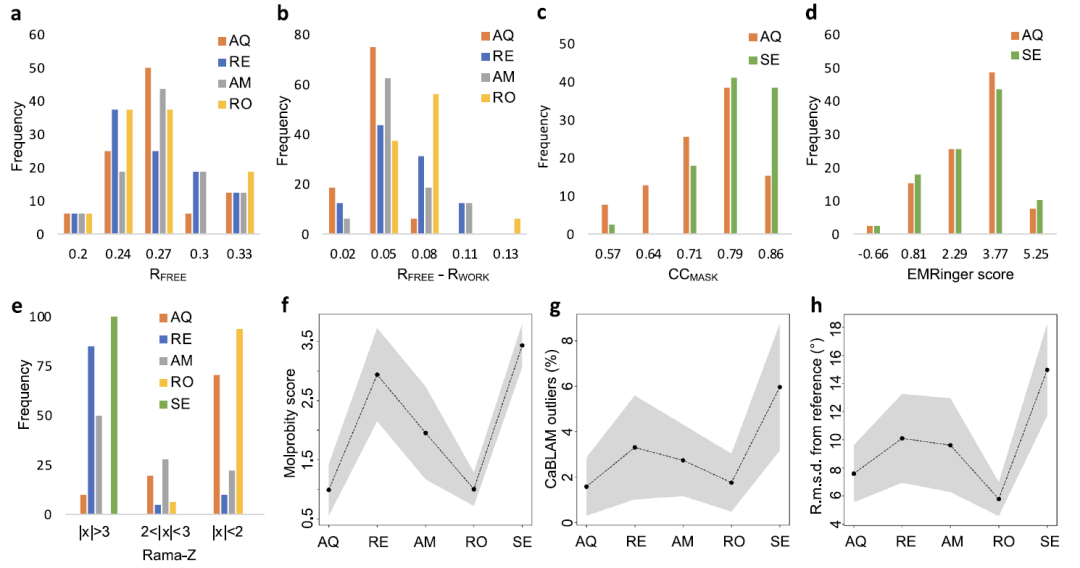
41回のクライオ電子顕微鏡観察と20回のX線モデル解析によって検証されたAQuaRefの局所構造は、2Åの精度で最適化された。
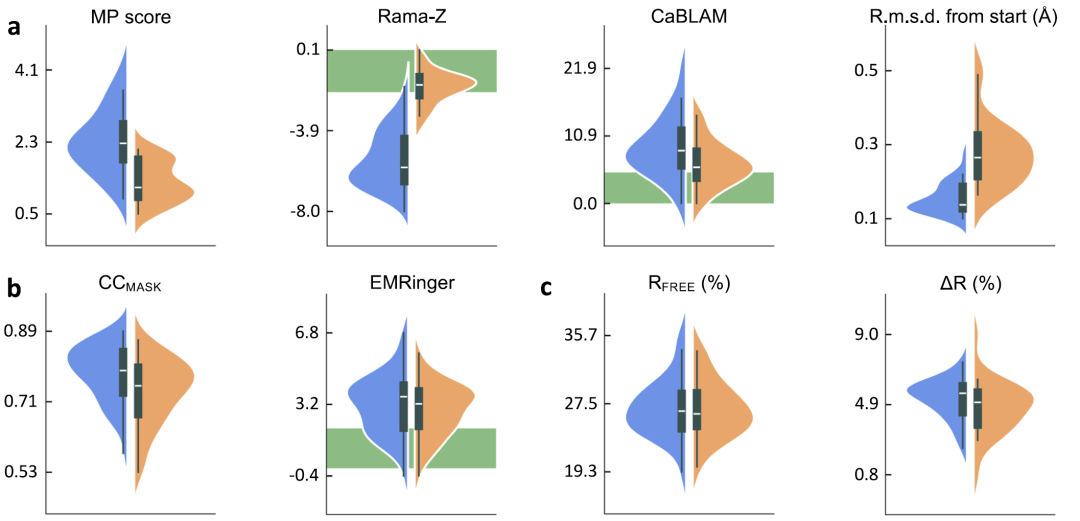
AQuaRefの性能を評価するために、研究者らは、41個のクライオ電子顕微鏡モデル、20個の低解像度X線モデル、および10個の超高解像度X線モデルを含むテストセットを構築した。61個の低解像度モデルすべてに、対応する高解像度の相同参照構造が付属しています。精製プロセスでは、比較のために3種類の制約が設定されました。AIMNet2量子制約(AQuaRef)、標準的な幾何学的制約、および標準的な制約に加えて水素結合や二次構造などの追加制約です。
結果は下の図に示されています。量子的に精緻化された低解像度モデルは、MolProbityスコアやラマチャンドランプロットZスコアなどの幾何学的指標において、従来の制約法を大幅に上回る性能を示す。一方、モデルの実験データへの適合度は概ね安定していた。X線構造については、過学習がわずかに減少した(RworkとRfreeの差が小さくなった)。クライオ電子顕微鏡構造については、CCmaskがわずかに減少したが、EMRingerスコアはほぼ変化しなかった。これらの結果と、幾何学的品質の全体的な向上を合わせると、モデルの過学習が軽減された可能性が示唆される。
標準的な制約に幾何学的制約を追加することでモデルの品質は向上するものの、AQuaRefはより妥当な形状を生成し、高解像度の参照モデルにより近いものとなる。場合によっては、標準的な制約と量子精密化構造との局所的な差が2Åに達することもある。
この研究では、AQuaRef をいくつかの主流の精製方法と比較しました。結果は下図に示されています。X 線データには AMBER、Rosetta、REFMAC5 が選択され、クライオ電子顕微鏡データには Servalcat が使用されました。全体として、AQuaRefはRfree性能がわずかに優れており、過学習の度合いも最も低い。Servalcatと比較すると、両者のEMRingerスコアは同程度だが、CCmaskスコアはServalcatの方がわずかに高い。
幾何学的品質の観点から言えば、AQuaRefはRosettaとほぼ同等の性能を発揮するが、REFMAC5やServalcatよりは大幅に優れた性能を示す。Rosettaは参照モデルとの全体的な適合度がわずかに高く、これは非勾配最適化戦略による収束半径の拡大に関連している可能性がある。さらに、AQuaRefとRosettaはどちらも妥当な水素結合幾何構造を生成でき、AMBERがそれに続く一方、REFMAC5とServalcatは基本的にこれらの詳細を正確に再現することができない。
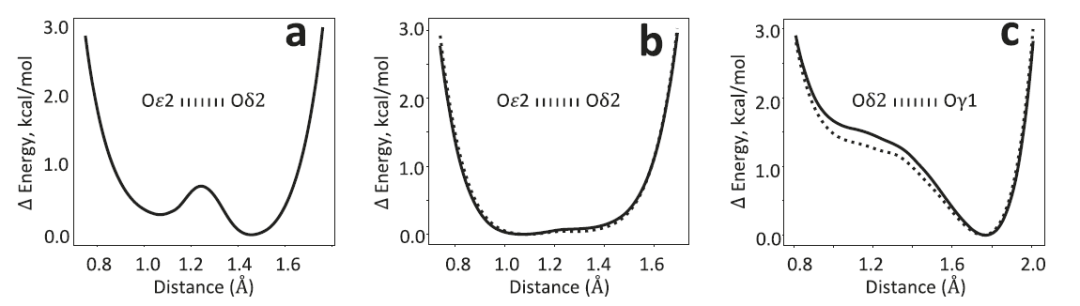
短い水素結合系に関するテストにおいて、研究者らはパーキンソン病関連タンパク質DJ-1とその相同タンパク質YajLを例として、AQuaRefがプロトン化状態を処理する能力を検証した。データベースの立体化学によって制約される従来の精製方法では、結合長が真の値からずれてしまうことがよくある。対称的な二重プロトン化構造をAQuaRef精密化の初期モデルとして使用した場合、得られたプロトン位置と結合幾何構造は、制約なし精密化の結果と一致する。従来の制約を追加すると、結合長はデータベース内の脱プロトン化標準値に近づきます。実験データが2 Åの分解能に切り詰められ、原子の詳細が大幅に減少した場合でも、AQuaRefは元の1.15 Åデータとほぼ同一の構造を復元できますが、従来の制約による精密化では真の配置からさらにずれてしまいます。AQuaRefは、DJ-1のD24残基のOδ2原子にプロトンを配置しますが、この結果はエネルギー計算と差分電子密度マップの両方によって裏付けられています。
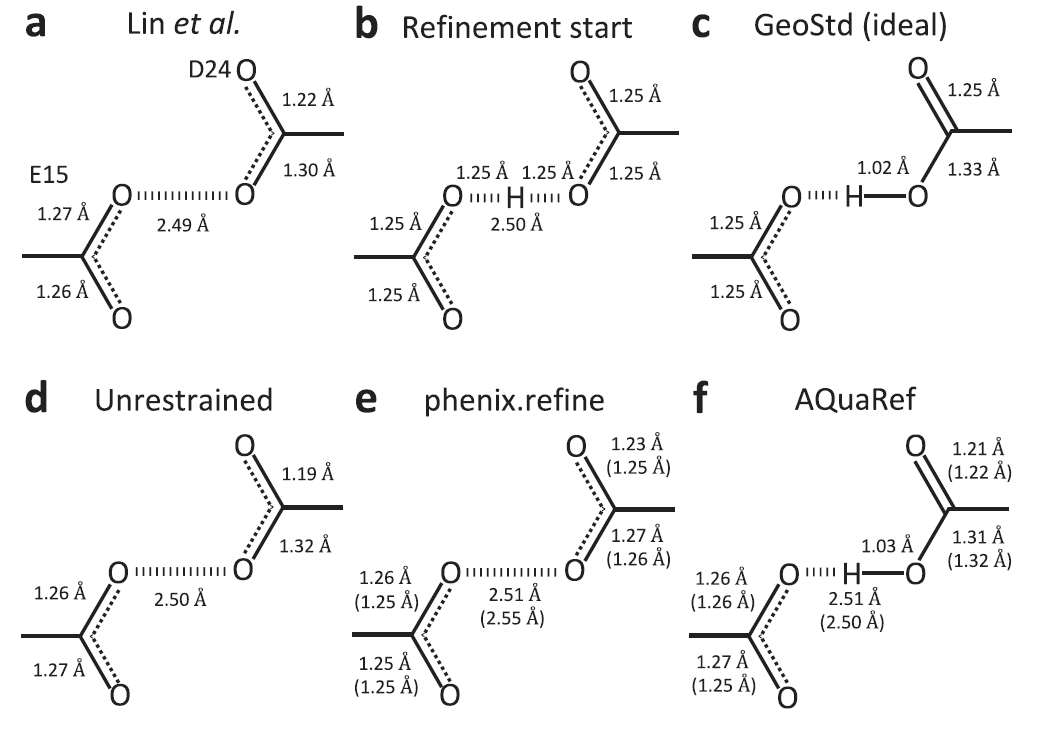
YajLタンパク質では、2つのE14/D23短水素結合に対するAQuaRef精密化の結果は、制約なし精密化と一致しており、プロトンがD23とE14の両方で共有され、典型的な低障壁水素結合特性を示している。これは、プロトンが主に単一の酸素原子上に位置するDJ-1の場合とは異なる。AIMNet2によって与えられたエネルギー分布は、比較的平坦なポテンシャルエネルギー面を示しており、実験データの制約の下でプロトンの位置を自由に調整できることを意味する。同時に、差分電子密度プロットは、水素原子付近で3σよりも有意に高いピークを示しており、この構造解釈をさらに裏付ける証拠となっている。
タンパク質量子精製分野における産学連携の画期的な進展
タンパク質の量子精密化、機械学習ポテンシャル関数の構築、原子モデル最適化といった最先端分野では、複数の研究チームがこの方向性を継続的に探求し、一連の進歩を遂げてきました。例えば、オックスフォード大学の研究チームが開発したニューラルネットワーク手法nn-tm fccは、ほぼ完全な量子力学的精度で、残基断片の高精度なポテンシャルエネルギー曲面モデルを構築することができる。エネルギー計算と原子間力計算の二乗平均平方根誤差は、それぞれ1.0 kcal/molおよび1.3 kcal/(mol·Å)以内に抑えられています。この方法を用いることで、代表的な15種類のタンパク質のエネルギー計算と原子間力計算をわずか10~100秒で完了させることができ、これは従来の量子力学計算よりも数千倍高速です。
論文タイトル:深層学習によるポテンシャルを用いたタンパク質構造予測の改善
論文リンク:https://www.nature.com/articles/s41586-019-1923-7
別のドイツの共同研究チームは、非可変反復戦略とIonQイオントラップ量子コンピューティングシステムを組み合わせたBF-DCQO量子アルゴリズムを提案した。12個のアミノ酸を含む3次元折り畳み問題の計算時間は、従来のGPUクラスタでは72時間かかっていたのが、約4.3分に短縮された。速度向上も千倍に達した。
論文タイトル:高次二値最適化のためのバイアス場デジタル化反断熱量子アルゴリズム
論文リンク:https://www.nature.com/articles/s42005-025-02270-3
全体として、量子力学的手法、機械学習ポテンシャル関数、および実験的な構造データの組み合わせは、生体高分子の構造を精密化するための新しい技術的アプローチを提供しており、低解像度構造モデリング、リガンド結合様式解析、機能部位研究などのシナリオにおいて、より安定した役割を果たすことが期待される。