Command Palette
Search for a command to run...
従来のマルチモーダル統合の限界を打破!MITは、細胞共有情報と細胞固有情報を明確に分離するAPOLLOフレームワークを提案します。
単一細胞生物学研究において、測定技術の急速な発展は、科学的探究の限界を絶えず拡大させています。マルチプレックスイメージング、単一細胞トランスクリプトームシーケンシング(scRNA-seq)、クロマチンオープンシーケンシング(scATAC-seq)、タンパク質存在量検出といった分野におけるブレークスルーにより、研究者は転写制御、クロマチン状態、タンパク質発現、形態構造など、多次元から単一細胞を包括的に観察することが可能になりました。これらのマルチモーダルデータは、生命のコードを様々なレベルから解釈し、それらを相互に統合することで、細胞の異質性を明らかにし、疾患メカニズムを探求するこれまでにない機会を提供します。
しかし、現在の分析方法では、このようなハイスループット データを処理する際に依然として大きな制限があります。主流の戦略では、各モードを個別に分析して比較することがよくありますが、これは非効率的であるだけでなく、モード間の深い相関関係を捉えることが難しくなります。別のタイプのアプローチでは、表現学習を通じてマルチモーダル データを同じ潜在空間に統合しますが、共有情報とモダリティ固有の情報が混同されることが多く、細胞機能に対する各次元の固有の寄与が不明瞭になります。
この問題は、scATAC-seq と scRNA-seq のペアデータの統合解析で特に顕著になります。従来の手法では、遺伝子発現と比較するために、クロマチンアクセシビリティを遺伝子レベルまで粗く粒度化することがよくあります。これは問題を単純化する一方で、クロマチンレベルの微細構造情報が失われる可能性があり、比較的均一な特性を持つデータタイプにしか適用できません。線形モデルや敵対的生成ネットワークといったより複雑な統合手法は、画像のような非構造化データへの適応が困難であったり、共通情報と固有情報の分離が不十分であったりするため、大規模バイオバンクにおけるマルチモーダルデータ分析の需要の高まりに対応できません。
したがって、単一細胞技術の継続的な進化とデータ規模の急速な拡大に伴い、共有情報とモダリティ固有の情報を明確に分離しながら、マルチモーダルデータを効率的かつ自動的に統合する方法が、単一細胞生物学が直面する中核的な課題となっています。
この課題に対処するため、MIT と ETH チューリッヒの共同研究チームは、APOLLO (潜在的最適化を通じて学習された部分的に重複する潜在空間を備えたオートエンコーダ) と呼ばれる汎用的なディープラーニング コンピューティング フレームワークを提案しました。このフレームワークは、共有情報とモダリティ固有の情報を明示的にモデル化することにより、細胞の状態とその制御ロジックをより包括的かつ正確に分析するための実現可能な技術的パスを提供します。
「部分的に共有されたマルチモーダル埋め込みが細胞状態の全体的な表現を学習する」と題された関連研究結果が Nature Computational Science に掲載されました。
研究のハイライト:
* 本研究では、マルチモーダルデータ内の「共有情報」と「モーダリティ固有の情報」を自動的かつ明示的に分離できる汎用的なディープラーニングフレームワークである APOLLO を提案します。
* APOLLO は、各モダリティにオートエンコーダーを装備し、2 段階のトレーニング戦略を採用することで、部分的に重複する潜在空間を学習し、複数のモダリティで一般的に捕捉される生物学的信号を効果的に識別および区別します。
* APOLLO は、タンパク質の細胞内局在の違いとさまざまな細胞区画の形態との関連性を明らかにすることができるため、分析範囲を純粋なオミクスデータから空間形態学の分野にまで拡張できます。

用紙のアドレス:
https://www.nature.com/articles/s43588-025-00948-w
弊社の公式 WeChat アカウントをフォローし、バックグラウンドで「APOLLO」と返信すると、完全な PDF を入手できます。
データセット: シーケンシングとイメージングを網羅した包括的な検証
APOLLO フレームワークのパフォーマンスを総合的に評価するために、この研究では、シーケンス技術とイメージング技術の両方を網羅した、公開されている複数のマルチモーダル単一細胞データセットを使用しました。
シーケンスデータに関しては、研究者らはまず、SHARE-seq テクノロジーで測定されたペアの単一細胞トランスクリプトーム (scRNA-seq) とクロマチン アクセシビリティ (scATAC-seq) のデータを使用して、トランスクリプトームとクロマチン アクセシビリティの両方で捕捉された遺伝子活動と、どちらか一方のモダリティのみで捕捉された遺伝子活動を APOLLO が自動的に識別して区別できるかどうかを検証しました。
次に、研究者らは、CITE-seq によって得られた scRNA-seq と細胞表面タンパク質存在量のデータをペアにして使用し、モデルのシーケンスデータへの適用可能性をさらにテストしました。CITE-seq データセットはマウスの脾臓とリンパ節から取得され、独立した実験的治療を受けた 2 つの野生型マウス サンプル グループが含まれています。これは、細胞タイプの識別能力を評価するために使用できるだけでなく、異なるマウスソースによって引き起こされる実験バッチ効果を明らかにすることもできます。
画像データに関しては、研究者らは、40人の患者から採取した32,345個のヒト末梢血単核細胞(PBMC)のマルチプレックス画像データセットを提示した。これらの細胞は、健常者、髄膜腫、神経膠腫、頭頸部腫瘍の4つの診断カテゴリーに分類された。各患者から、異なる抗体の組み合わせに基づいて2セットの画像データが収集された。1セット目はDAPIを用いてクロマチンを標識し、CD4、CD8、CD16抗体染色と組み合わせた。もう1セット目はDAPI染色を用い、ラミン、CD3、γH2AX抗体染色と組み合わせた。
このデータセットを使用したテストでは、APOLLO は、クロマチン構造とタンパク質の局在に関する 2 つのモダリティ間で共有される細胞状態情報だけでなく、単一のモダリティによってのみ捕捉される形態学的特徴も識別できます。さらに、微小管や小胞体などの追加の細胞染色マーカーを組み合わせることで、この研究では、ヒトタンパク質アトラス (HPA) からの複数の画像データも使用し、APOLLO を使用してタンパク質の細胞内局在の違いと異なる細胞区画の形態との関連性を明らかにできることを実証しました。
APOLLOモデル: 潜在最適化戦略を採用したオートエンコーダ
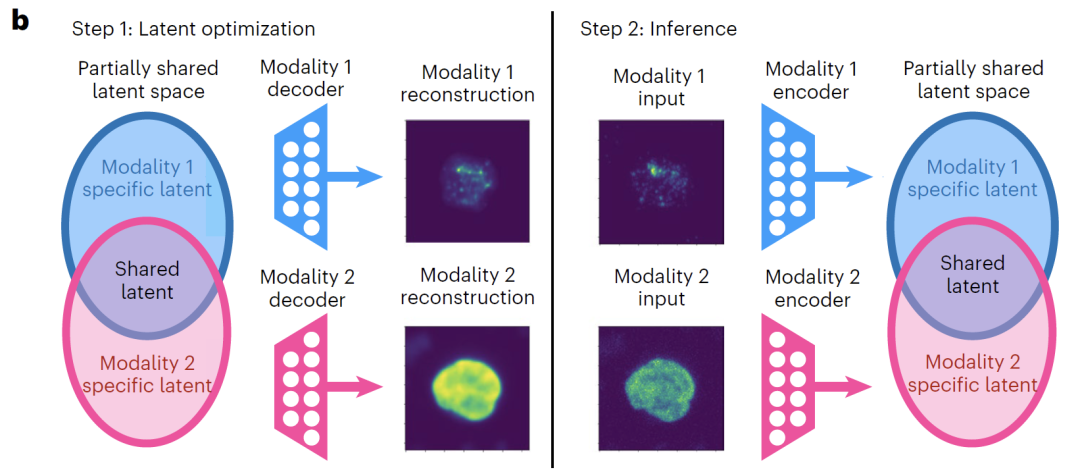
既存のマルチモーダル統合手法に共通する問題、すなわち共通情報とモダリティ固有の情報が混同されるという問題に対処するため、本研究ではAPOLLOフレームワークを提案する。このフレームワークは、潜在最適化を用いて部分的に重複する潜在空間においてオートエンコーダを学習し、複数のモダリティ間で共通情報とモダリティ固有の情報を自動学習し、効果的に分離することを目指す。従来のオートエンコーダはすべての潜在次元を均一にアラインメントさせるが、APOLLOフレームワークは、APOLLO は、潜在的な次元の一部に対してのみクロスモーダルアライメントを実行し、残りの次元を各モーダリティに固有の情報のために予約することで、モデル設計において共有情報と固有の情報を明確に分離します。
モデルアーキテクチャの観点から言えば、APOLLO には各データ モダリティ用のオートエンコーダが装備されており、タスクの必要に応じて追加のデコーダを導入できます。エンコーダとデコーダは、特定のモダリティに適応したニューラルネットワーク構造を採用しています。例えば、画像データには畳み込みネットワーク、遺伝子発現データには全結合ネットワークを用いることで、各モダリティのデータ特性を完全に捉えることができます。潜在空間は、共有潜在特徴とモダリティ固有の潜在特徴の2つの部分に明示的に分割されます。共有潜在空間の次元は、通常、モダリティ間の共通情報を十分に表現するために、モダリティ固有の空間の次元よりもはるかに大きく設定されます。
下の図に示すように、APOLLO のトレーニング プロセスは 2 つのステップで構成されます。最初のステップでは、潜在空間を同時に更新しながら、各モダリティのデコーダーをトレーニングすることに重点を置きます。主な目的は、デコーダーが潜在空間から入力データを正確に再構成できるようにすることです。タスクが共有情報表現の強化とクロスモーダル予測の達成を必要とする場合、共有潜在空間を各モーダリティにそれぞれマッピングする2つの追加デコーダーが導入され、再構成損失を最小化することで学習が完了します。
2 番目のステップは、モダリティ固有のエンコーダーをトレーニングすることです。各データモードは対応する潜在空間にマッピングされます。平均二乗誤差を最小化することで、学習に関与しないサンプルの潜在空間への埋め込みを推定し、モデルの優れた汎化能力を確保します。
モデル検証のため、この研究ではまず、実際の基礎構造がわかっている 5 つのシミュレートされたデータセットで APOLLO の分離パフォーマンスをテストしました。結果は、共有された潜在的特徴と特定の潜在的特徴間の依存関係に関係なく、モデルが安定したパフォーマンスを維持できることを示しています。実際のデータによるさらなる検証により、APOLLO の部分的な情報共有の明示的な学習により、マルチモーダル情報を分離できるだけでなく、クロマチンイメージングから検出されていないタンパク質を予測するなど、正確なクロスモーダル予測も実現できることが示されました。
全体として、APOLLO は、部分的に共有された潜在空間を学習することにより、マルチモーダル データセット内の共有情報とモダリティ固有の情報を効果的に分離して解釈し、生物学的メカニズムを解明するための一般的なフレームワークを提供します。
従来のマルチモーダル統合フレームワークを超えて、細胞の状態をより包括的に理解します。
APOLLO モデルの普遍性と中核的利点を総合的に評価するために、ペアシーケンスデータの統合、クロマチンとタンパク質のイメージングの統合、クロスモーダル予測、形態学的特徴の認識、およびタンパク質の細胞内局在の探査という 5 つの方向を中心に一連の実験が設計されました。
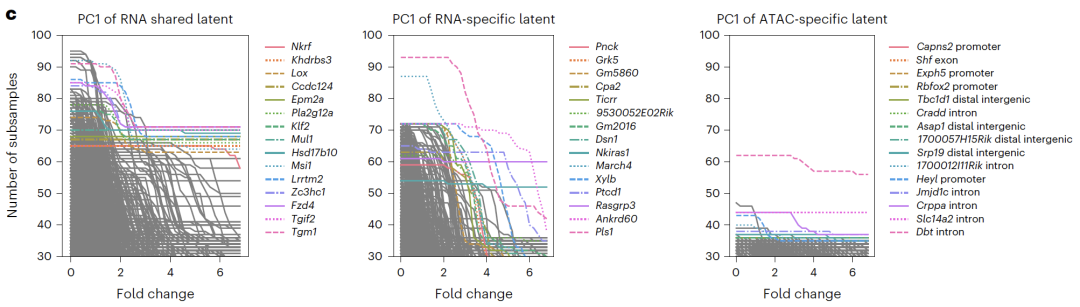
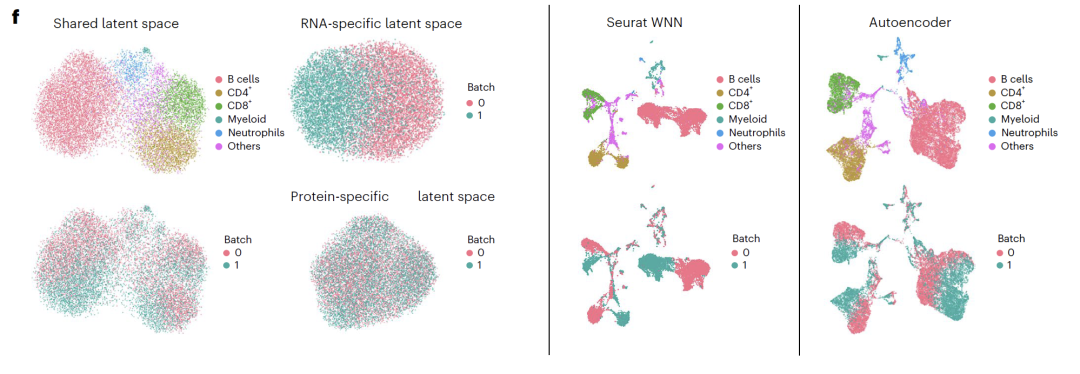
ペアシーケンスデータ統合SHARE-seq の実験では、共有空間にモダリティ固有の空間を追加することで細胞タイプの分類精度が大幅に向上することが示され、特定の空間が共有空間に含まれていない生物学的情報を取得できることが証明されました。
空間的解釈の可能性から、RNA特異的空間には細胞周期関連遺伝子が豊富に存在し、ATAC特異的空間には転写制御に関連するオープンクロマチン領域が豊富に存在し、共有空間には既知の転写因子と制御経路が豊富に存在することが明らかになり、分離結果の生物学的意義が検証された。CITE-seq実験では、APOLLO は、細胞タイプとバッチ効果を共有スペースと RNA 固有のスペースに分離することに成功しました。既存の統合方法ではこのような分離は実現できないため、シーケンス データを統合する際のこのモデルの独自の利点が強調されます。
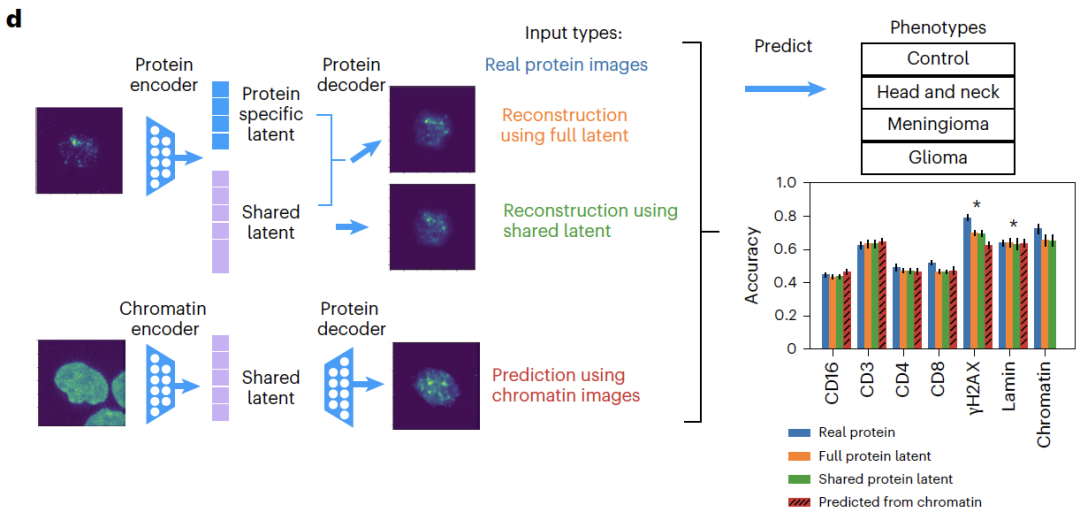
画像データに関しては、APOLLO は、トレーニングに参加していない患者の細胞画像を正確に再構築できます。クロマチンから検出されないタンパク質を予測するクロスモーダルタスクにおいて、APOLLO は従来の画像修復手法を大幅に上回ります。下流の表現型分類では、予測されたタンパク質イメージングに基づく分類精度が実際のイメージングの精度と同等であり、CD3タンパク質が最高の予測性能を示し、予測結果が生物学的発見に効果的に使用できることが確認されました。
形態学的特徴認識タスクでは、共有空間は主にクロマチンの形態学的特徴(核面積やヘテロクロマチン体積など)を捉えており、γH2AXフォーカルカウントなどのタンパク質特異的な特徴は、対応する特定の空間にのみ存在します。特徴除去実験では、この特徴を除去すると表現型分類の精度が大幅に低下することが示され、分離の精度がさらに検証されました。

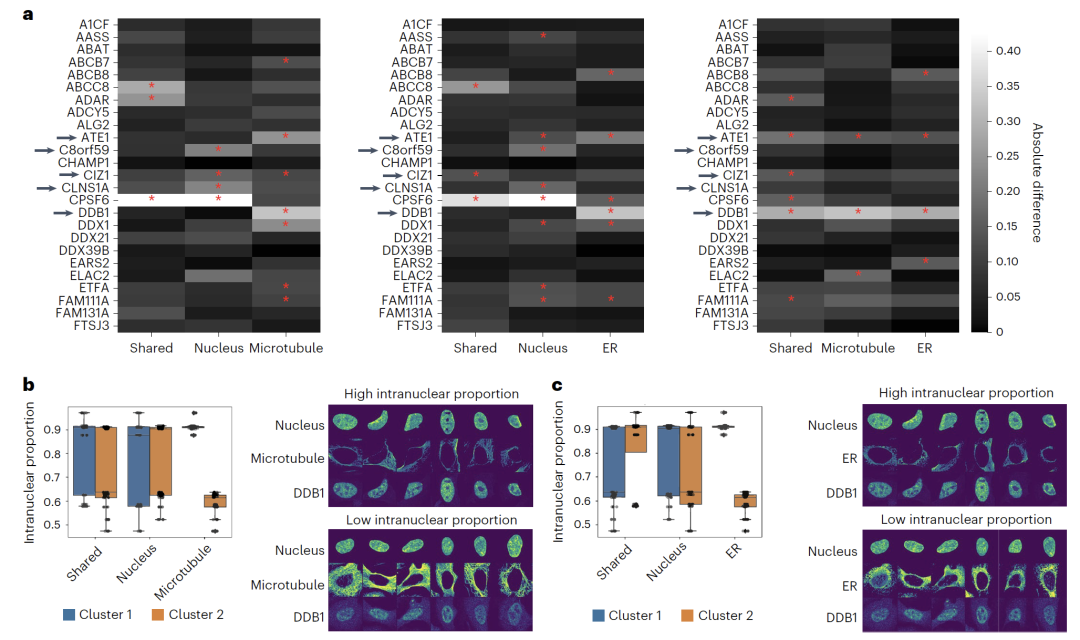
タンパク質の細胞内局在の研究においてAPOLLOをU2OS細胞イメージングデータに適用することで、核腔内のタンパク質局在の違いが、細胞コンパートメントの特性によって捉えられることが明らかになりました。例えば、DDB1の核局在は小胞体と微小管の形態と関連しているのに対し、CLNS1Aは核の形態のみと関連しています。この結果は…このモデルはさまざまな画像の組み合わせに拡張でき、タンパク質の局在と細胞形態の関係を理解するための新しい視点を提供します。
単一細胞マルチモーダルデータ統合の実装
単一細胞マルチモーダルデータの統合は、細胞の異質性を分析し、疾患のメカニズムを明らかにし、精密医療の発展を促進するための中核的な技術的方向性になりつつあり、世界中の学術界から広く注目を集めています。
たとえば、ケンブリッジ大学バブラム研究所のピーター・ラグ・ガン氏のチームによって開発された scMTR-seq テクノロジーは...初めて、6 つのヒストン修飾とトランスクリプトーム全体の同時捕捉が単一細胞レベルで達成されました。彼らは、10年間続いたエピジェネティクス研究の分野における技術的なボトルネックを克服した。
論文のタイトル:
scMTR-seqを用いた単一細胞における複数のヒストン修飾とトランスクリプトームの組み合わせプロファイリング
論文リンク:
https://www.science.org/doi/10.1126/sciadv.adu3308
スタンフォード大学の研究チームが提案した CellFuse フレームワークは、教師あり対照学習に基づいて共有埋め込み空間を構築し、機能の重複が制限されたマルチモーダル統合シナリオ向けに特別に設計されています。正確な細胞タイプの予測と、モダリティや実験条件間のシームレスな統合を実現できます。健康な PBMC、骨髄、リンパ腫の CAR-T 療法、腫瘍組織など、複数のデータセットでのテストにより、このフレームワークは統合品質と運用効率の両方において既存の方法よりも優れていることが実証されています。
論文のタイトル:
CellFuseは単一細胞と空間プロテオミクスデータのマルチモーダル統合を可能にします
論文リンク:
一方、世界をリードするバイオテクノロジー企業やヘルスケア企業は、臨床応用、医薬品開発、精密医療といった中核分野に重点を置き、シングルセル・マルチモーダルデータ統合技術の導入を加速させ、最先端研究の実用化を推進しています。ドイツのBioNTech社は、この技術を腫瘍免疫療法と個別化ワクチン開発に応用しています。シングルセルRNAシーケンシング、タンパク質発現プロファイリング、空間トランスクリプトミクスデータを統合することで、腫瘍微小環境における細胞の不均一性を精密に解析し、主要な免疫細胞サブタイプと関連バイオマーカーを特定することで、個別化腫瘍ワクチンの設計と最適化のためのコアデータを提供し、ワクチンのターゲティングと有効性を大幅に向上させています。
マルチモーダル統合技術の継続的な進歩により、単一細胞レベルでの生命の解読が最終的にはビジョンから現実へと移行し、精密医療の未来にさらに強い推進力を注入することが予測されます。