Command Palette
Search for a command to run...
ワシントン大学とマイクロソフトなどは、14,000件の実際のデータセットに基づいて、腫瘍免疫微小環境のパノラマアトラスを作成するためにGigaTIMEを提案しました。
がんの進化の過程において、腫瘍免疫微小環境はがん細胞の増殖、浸潤、転移を支配するだけでなく、治療反応や患者の最終的な予後にも深く影響を及ぼします。これはがん細胞の「独演」ではなく、非常に動的な生態系です。免疫細胞、線維芽細胞、内皮細胞などが複雑に絡み合い、相互作用しながら、構造と機能が再構築された細胞外マトリックスに共存し、精密で複雑な病理学的ネットワークを形成します。
このネットワークを解読する鍵は、細胞の機能状態と相互作用を理解することにあり、特定のタンパク質の活性化レベルは重要な「分子コード」です。伝統的に、免疫組織化学 (IHC) は、タンパク質の局在を視覚的に確認できるため、コードを解読するための古典的なツールとなっています。例えば、PD-L1染色は、免疫チェックポイントの状態を特定し、免疫療法の有効性を予測するために広く利用されてきました。しかし、IHCでは一度に1つのタンパク質の情報しか取得できないため、複数のタンパク質が共存する真の生態を再構築することが困難であり、これが腫瘍と免疫細胞の対話メカニズムをより深く理解する上で大きなボトルネックとなっています。
この限界を克服するために、マルチプレックス免疫蛍光(mIF)技術が開発されました。この技術は、単一の組織切片における複数のタンパク質の空間分布を同時に提示することができ、組織構造の文脈情報を完全に保持します。しかし、この技術は高価でプロセスが複雑であり、染色、画像化、分析のすべてに非常に時間がかかります。これにより、大規模なデータの蓄積が困難になり、臨床への応用が妨げられます。
対照的に、H&E染色切片は臨床現場で広く入手可能で、安価です。タンパク質の活性を直接明らかにすることはできませんが、組織の全体構造と細胞形態の詳細を完全に保存しています。切片に隠された特徴は、細胞の機能状態を間接的に反映している可能性がありますが、これらの微妙で複雑なパターンは、しばしば人間の視覚の限界を超えています。
近年、人工知能技術の飛躍的進歩は新たな可能性をもたらしました。膨大な病理画像の事前学習を通じて、AIは強力な視覚分析と特徴マイニング能力を発揮しました。これは重要な疑問へと繋がります。AIは、これまで高価なmIF技術を用いて取得する必要があった、容易に入手できるH&E画像からタンパク質活性化情報を「解読」できるのでしょうか?
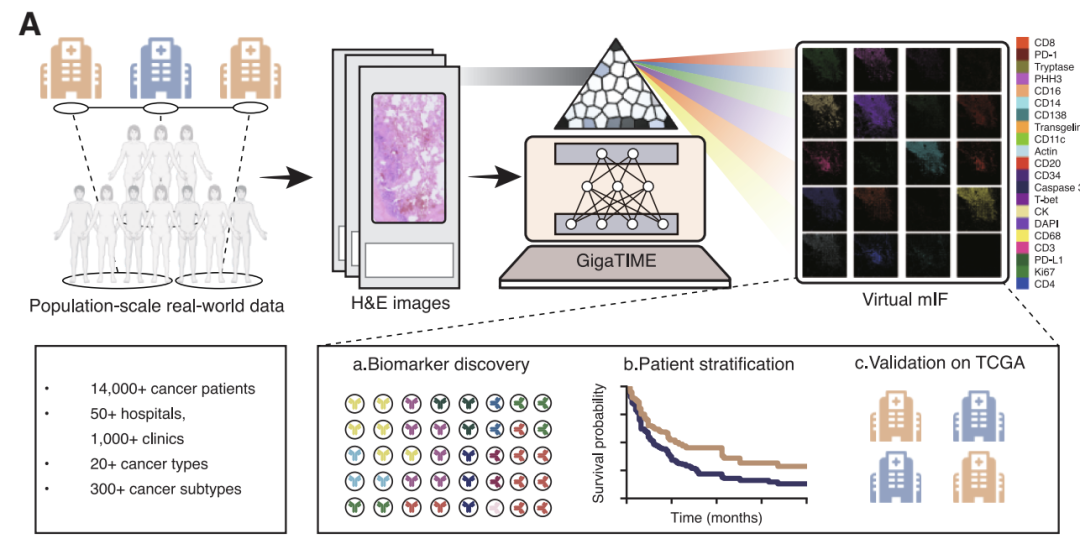
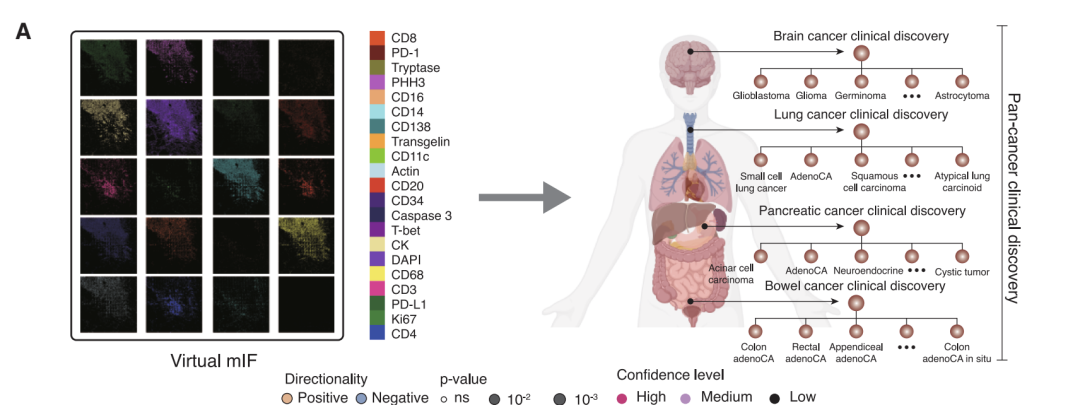
この考え方に基づいて、Microsoft Research、ワシントン大学、Providence Genomics からなる研究チームが、マルチモーダル人工知能フレームワーク GigaTIME を提案しました。高度なマルチモーダル学習技術を活用することで、従来のH&Eスライス画像から仮想mIFマップを生成することができます。研究チームは、米国プロビデンス医療センターの14,000人以上の癌患者コホートにこの技術を適用し、24種類の癌と306のサブタイプを網羅しました。最終的に約30万枚の仮想mIF画像を生成し、大規模かつ多様な集団における腫瘍免疫微小環境の体系的なモデリングを実現しました。
「マルチモーダル AI が腫瘍微小環境モデリングのための仮想集団を生成する」と題された関連研究結果が Cell に掲載されました。
研究のハイライト:
* GigaTIME は、マルチモーダル AI を使用して H&E 病理スライドを空間プロテオミクス データに変換し、通常の H&E スライドから細胞の状態を含む仮想集団を生成します。
* 大規模な臨床発見と患者の層別化をサポートし、新しい空間的および組み合わせ的なタンパク質活性化パターンを明らかにします。

用紙のアドレス:https://www.cell.com/cell/fulltext/S0092-8674(25)01312-1
弊社の公式 WeChat アカウントをフォローし、バックグラウンドで「Multiple Immunizations」と返信すると、完全な PDF を入手できます。
AIフロンティアに関するその他の論文:
https://hyper.ai/papers
データセット: トレーニングからアプリケーションまでの完全なクローズドループの構築
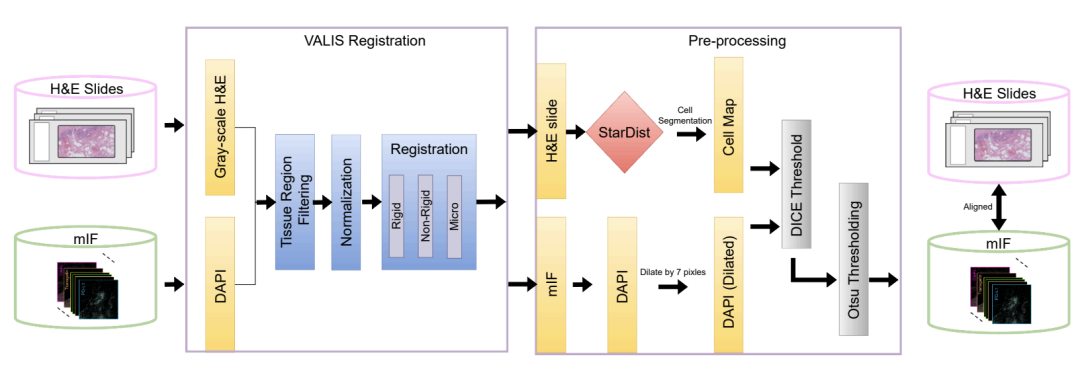
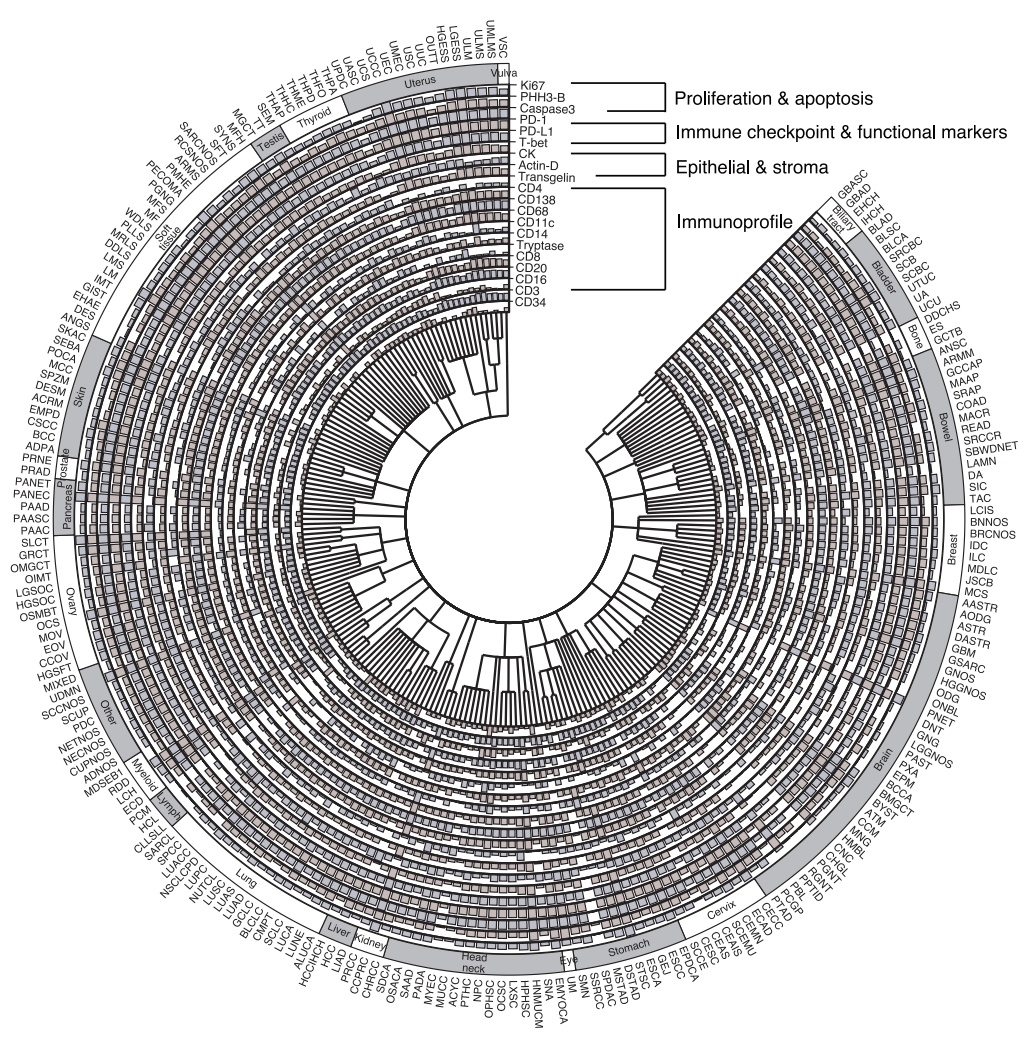
モデルの学習には、まず根本的な矛盾に対処する必要があります。臨床現場で広く利用され低コストなH&E染色はタンパク質の活性を直接明らかにすることはできませんが、複数のタンパク質間の空間的関係を明らかにできるmIF技術は高価で複雑であるため、大規模な導入は困難です。これら2つの画像技術を連携させたAIモデルを構築するには、研究チームは COMET プラットフォームを使用して、21 個の H&E 染色切片から 441 個の mIF 画像を収集しました。下図に示すように、これらの画像は、DAPIやPHH3などの核タンパク質、CD4やCD11cなどの表面タンパク質、CD68などの細胞質タンパク質に至るまで、合計21の主要バイオマーカーを網羅しています。これらは、腫瘍微小環境における免疫細胞の構成、機能状態、および活性を解析するための重要な証拠となります。

ペア画像を取得した後、より大きな課題は、そこから高品質のトレーニングデータを抽出することです。この目的のために、チームは下図に示すように、厳密な処理ワークフローを設計しました。まず、VALISツールを使用してH&E画像とmIF画像をピクセルレベルで正確に位置合わせします。次に、StarDistアルゴリズムを使用して画像内の各細胞を識別およびセグメント化します。最後に、Dice係数に基づいて、位置合わせ品質が最も高い画像領域を選択します。
多層的な品質管理を通じて研究チームは、4,000 万個の細胞を含む初期データから 1,000 万個の高品質細胞を選択し、それをトレーニング セット、検証セット、独立テスト セットに分割しました。さらに、本研究では、組織マイクロアレイから得られた乳がんおよび脳腫瘍のサンプルを外部検証セットとして導入しました。これらのサンプルは、組織構造と形態においてトレーニングデータとは大きく異なっていました。トレーニングデータでは大きな連続した組織切片が見られましたが、これらのサンプルは空白領域で区切られた小さな円筒形の組織ブロックとして現れました。これにより、新しいサンプルタイプや未知のがん種に直面した際のモデルの汎化能力を効果的に検証することができました。
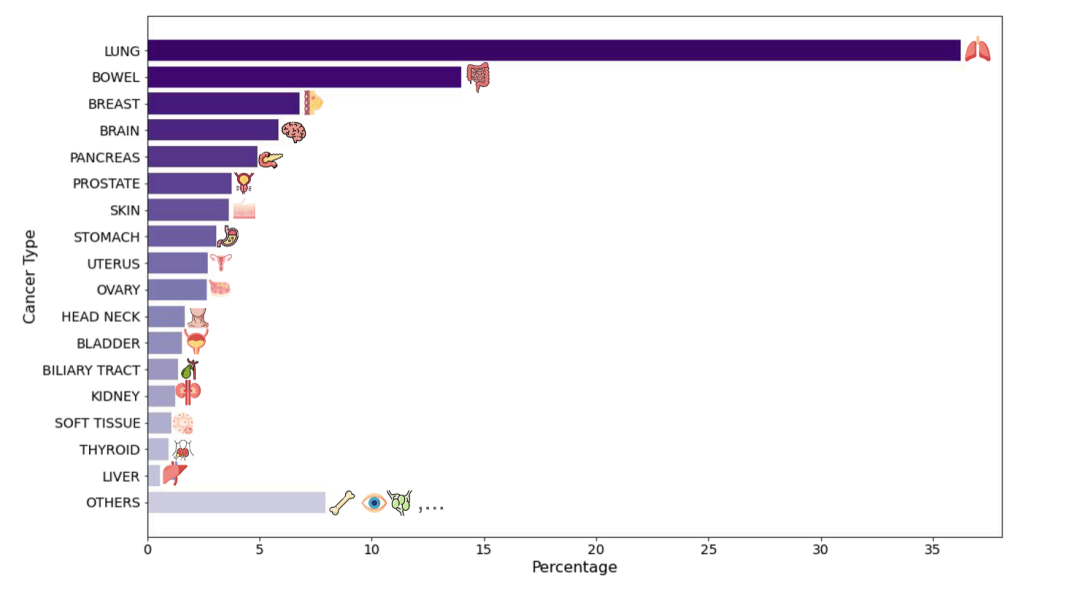
モデル応用レベルでは、この研究では 2 つの大規模かつ補完的な仮想人口コホートを構築しました。最初のコホートは、米国を拠点とする医療グループであるプロビデンス・ヘルスの臨床ネットワークから提供され、51の病院と1,000以上のクリニックに所属する14,256人のがん患者のH&Eスライス画像が含まれています。24の主要ながん種と306のサブタイプを網羅し、ゲノムバイオマーカー、病理学的ステージ分類、生存追跡調査などの豊富な臨床情報を統合しています。このデータセットの独自の価値は、その現実世界の特性にあります。早期から後期まで、あらゆる病期を網羅する非常に多様な患者集団であり、臨床現場の複雑さを現実的に反映しています。
2 番目のコホートは、Cancer Genome Atlas (TCGA) 公開データベースから取得されました。本研究には、主に早期段階の未治療外科手術症例から得られた10,200枚のH&Eスライドが含まれました。これら2つのコホートは、患者の起源、病期、臨床状況において非常に対照的でした。この異なるデザインは、モデルの信頼性と一般化可能性を検証するための優れた条件を提供しました。このように多様なデータセット全体にわたって一貫性のある堅牢な生物学的結論が得られたことから、モデルの幅広い臨床的可能性が強く示唆されます。
GigaTIME: 形態と機能のインテリジェントな橋を構築する
GigaTIMEモデルは、腫瘍免疫微小環境研究における主要なボトルネックに直接対処します。高コストで低スループットのmIF技術は普及が難しく、日常的な臨床H&E染色画像ではタンパク質の機能活性を直接反映できないという問題です。このモデルは、人工知能を用いてH&E画像から学習・仮想mIF画像を生成することで、集団スケールにおける腫瘍免疫微小環境の低コストかつ体系的な解析を実現する実現可能な道筋を提供します。
このモデルは、慎重に設計されたパッチワーク エンコーダー/デコーダー フレームワークを採用しており、その中核はネストされた U 字型ネットワーク上に構築されています。このアーキテクチャの利点は、画像の微細な局所的特徴と全体的な組織構造の両方を同時に捉えることができる点にあります。具体的には、ネットワークのエンコーダ部は、入力となる256×256ピクセルのH&E画像パッチから、畳み込みとダウンサンプリング処理によって多段階の特徴表現を抽出します。デコーダ部は、これらの抽象的な特徴を、アップサンプリングと特徴融合によって空間分解能を備えた仮想mIF画像に再構成します。この設計により、モデルは個々の細胞の微細な形態と細胞集団の組織パターンの両方に焦点を当てることができます。
出力レベルでは、モデルの設計は生物学的問題に対する深い考慮を反映しています。GigaTIME は、21 個のプリセットされたタンパク質チャネルごとに、入力画像内のすべてのピクセルに対してバイナリ分類予測を実行します。このシステムは、特定のタンパク質が特定の場所で活性化されているかどうかを判定し、最終的にピクセルレベルのタンパク質活性マップを生成します。これらの局所予測はシームレスにつなぎ合わせることで、組織切片全体の仮想mIF画像を再構成できます。これにより、腫瘍領域における特定タンパク質の活性化密度や空間分布パターンなど、様々な定量指標の算出が可能となり、その後のハイスループット解析や臨床相関研究のための確固たるデータ基盤を提供します。
効果的なモデル学習を確実にするために、トレーニング戦略は体系的に最適化されています。この損失関数は、ダイス損失とバイナリクロスエントロピー損失を巧みに組み合わせています。前者は、予測されたアクティブ領域とグラウンドトゥルース領域の空間輪郭における全体的な一貫性を確保することに焦点を当て、後者は各ピクセルの分類精度の向上に焦点を当てています。この2つの相乗効果により、グローバルな空間パターンの正確な再構築と詳細レベルでの信頼性の両方が確保されます。モデルは、8基のNVIDIA A100 GPU上で、バッチサイズ16、学習率0.0001で250エポック学習されました。すべての主要なハイパーパラメータは、検証セットの結果に基づくシステムデバッグを通じて決定されました。
モデルの成功は高品質のトレーニング データに大きく依存することを強調することが特に重要です。研究チームは、厳密な画像登録、細胞分割、品質管理手順を採用しました。トレーニング用に大量の初期データから 1,000 万個の高品質セルが選択され、モデルが表面的な統計的規則性やノイズの多いパターンではなく、堅牢で信頼性が高く、生物学的に意味のあるクロスモーダル マッピングを学習することが保証されます。
約30万枚の仮想画像に基づく大規模な調査結果:GigaTIMEが1,234の臨床的関連性を明らかに
GigaTIME の性能と価値を総合的に評価するために、研究チームは技術検証と臨床所見という 2 つの側面から体系的な評価スキームを設計しました。
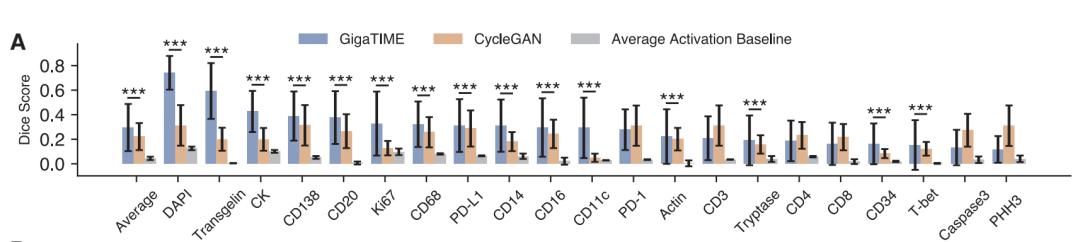
技術的な検証に関しては、この研究では、ピクセル、セル、スライスの 3 つのレベルでモデルの画像変換機能を評価しました。ピクセルレベルでは、GigaTIMEは21個のタンパク質チャネルのうち15個においてベースラインモデルCycleGANを大幅に上回ります。例えば、DAPIチャネルでは、GigaTIMEのDice係数は0.72となり、単純統計ベースラインの0.12を大きく上回ります。
細胞レベルでは、GigaTIME は DAPI チャネルで 0.59 の相関を達成しましたが、CycleGAN は 0.03 にしか達せず、ランダムなレベルに近づきました。
スライスレベルでGigaTIME の DAPI チャネル相関係数は 0.98 と高く、全チャネルの平均は 0.56 ですが、CycleGAN は 0 に近くなっています。これらの結果は、高品質のペアデータに基づく教師ありトレーニングが、正確なクロスモーダル変換に不可欠であることを示しています。
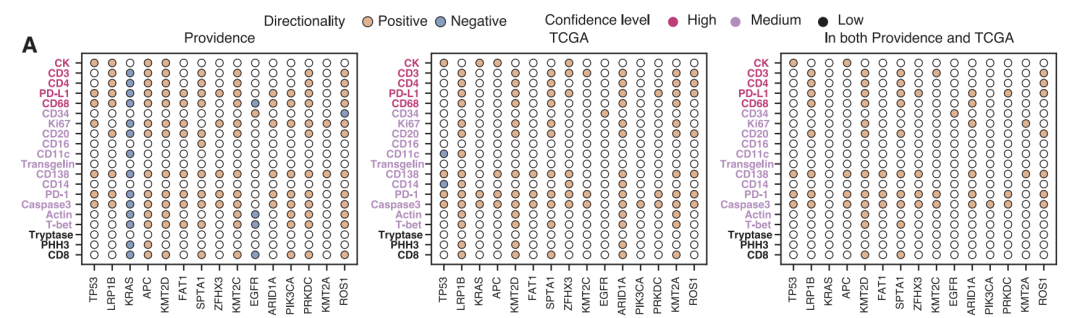
臨床的知見に関しては、この研究では 14,256 人の患者からの約 300,000 枚の仮想 mIF 画像が使用されました。仮想タンパク質発現と 20 の臨床バイオマーカーとの関連性を体系的に分析しました。厳密な統計的検定と複数の補正を行った結果、全がん、がんの種類、がんのサブタイプの 3 つのレベルにわたって合計 1,234 件の有意な関連性が特定されました。
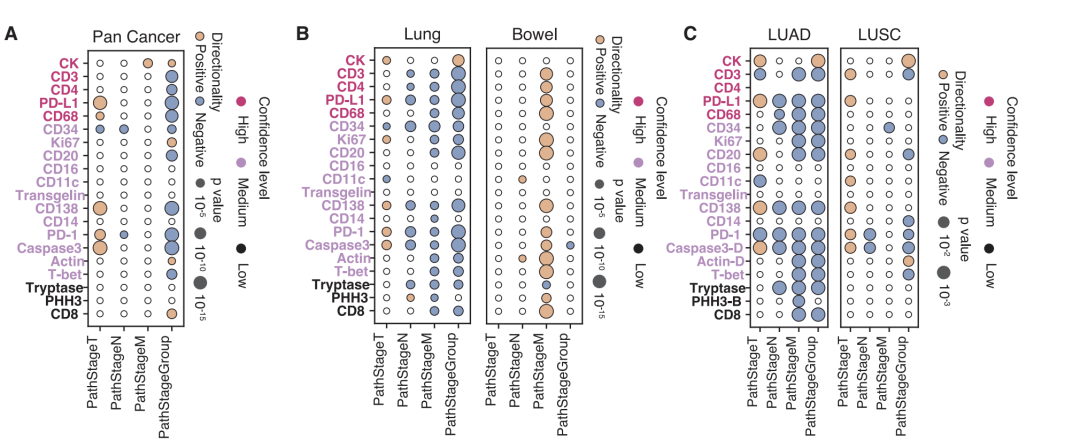
がん全体にわたる解析で特定された 175 の関連性のうち、腫瘍の変異負荷の高さとマイクロサテライト不安定性の高さは、複数の免疫浸潤マーカー (CD138、CD20、CD68、および CD4) の活性化の増強と有意に関連しており、抗原駆動型免疫活性化メカニズムと一致しています。新たな手がかりも発見されました。KMT2D 変異は免疫マーカーと強い正の相関を示し、免疫浸潤を促進する可能性があることを示唆しています。一方、KRAS 変異は負の相関を示し、免疫拒絶表現型を反映しています。特定のがんの種類とサブタイプでは、モデルによって多数の特定の関連性が明らかになりました。たとえば、脳腫瘍における T-bet 変異と TP53 変異の強い相関は、がん全体レベルでは検出されませんでしたが、これは中枢神経系の独特な免疫微小環境に関係している可能性があります。肺がんのサブタイプ解析により、肺腺がんにおける PRKDC 変異は扁平上皮がんよりも免疫応答マーカーと強く関連していることが示され、組織学的背景と併せてデータを解釈することの重要性が確認されました。
この研究では、臨床結果における仮想データの価値も検証されました。解析の結果、原発腫瘍の大きさ(Tステージ)と免疫チェックポイントおよび浸潤マーカーとの間に正の相関が認められましたが、進行期ではこの相関は逆転しており、進行腫瘍は主に他の免疫逃避機構によって引き起こされている可能性を示唆しています。生存解析では、21のパスウェイすべてを統合した複合解析は、患者層別化において単一タンパク質解析よりも優れており、複数のパラメータを組み合わせた解析の価値を十分に示しています。
信頼性を確保するため、すべての主要な知見はTCGAとは独立したコホートで検証されました。2つの集団間で起源と臨床的特徴に大きな違いがあったにもかかわらず、主要な知見は非常に一貫していました(がんサブタイプレベルでのスピアマン相関係数は0.88)。共通して特定された 80 件の有意な関連性は、極めて高い統計的エンリッチメントを示しました (p < 2×10⁻⁹)。一方、プロビデンス ヘルスの仮想人口は、全がんレベルで TCGA よりも 331 件多い有意な関連性を示し、大規模な現実世界のデータの独自の価値を浮き彫りにしました。
探索的解析により、複雑な空間パターンの価値も明らかになりました。エントロピー、信号対雑音比、シャープネスといった指標は、それぞれ89、63、79のタンパク質-バイオマーカーペアにおいて、単純な活性化密度よりも優れた結果を示しました。また、この研究ではタンパク質間の相乗効果も発見されました。CD138 と CD68 の組み合わせは、20 種類のバイオマーカーの予測において単一のタンパク質よりも優れた結果を示しました。これらの違いのうち 13 は統計的に有意であり、形質細胞とマクロファージが抗体を介したメカニズムを通じて腫瘍と戦うために協力している可能性があることを示唆しています。
AI活用:仮想タンパク質マップからがん研究の新たなフロンティアへ
AIを用いて日常的な病理標本から仮想プロテオミクス画像を生成することは、デジタル病理学と計算生物学におけるイノベーションの中核を成しています。この方向性は世界中のトップクラスの学術機関の注目を集め、バイオテクノロジー企業による商業的実践も促進しています。
学問の世界では、スタンフォード大学がNature Medicineに発表したHEXモデル819,000枚の画像ブロックを用いて学習させたこのシステムは、40種類のバイオマーカーの空間的発現を予測することができ、GigaTIMEよりも広範なタンパク質カバレッジを実証しています。カリフォルニア大学サンフランシスコ校がScience Translational Medicine誌に発表したDeepHemeシステムは、約50,000件の高品質な多施設データセットに基づき、23種類の骨髄細胞を正確に分類することに成功し、血液疾患診断の自動化に向けたパラダイムを提供しています。
業界において、Reveal Biosciences はビル&メリンダ・ゲイツ財団の支援を受けています。病理画像から「デジタルバイオマーカー」を抽出するプラットフォームを開発します。グローバルヘルス研究の加速。もう一つのアプローチは、サンプルと試薬の消費量を大幅に削減するMicronit社のマイクロ流体デバイスのようなハードウェアイノベーションを通じてコストを削減することです。Optellum社のFDA承認済み肺結節診断プラットフォームは、日常的なデータからより深い特徴を抽出し、臨床意思決定に役立てるための商業的パラダイムと規制上の先例を提供します。
GigaTIME はこの分野における重要なマイルストーンです。これは、腫瘍免疫微小環境の研究におけるマルチモーダル AI の大きな可能性を示すだけでなく、その後の研究に再利用可能な技術的フレームワークとデータ リソースも提供します。今後の開発は、「仮想現実」データ生成機能と低コストの検出技術の複合的な進歩にかかっており、最終的には腫瘍の複雑性を理解し、精密医療を加速するための革新的なツールがもたらされるでしょう。
参考リンク:
1.https://mp.weixin.qq.com/s/AsqSemP3idCbIJ7xQ3gXGg
2.https://mp.weixin.qq.com/s/umg-UrMm6Qe-R-MbLpLZOQ








