Command Palette
Search for a command to run...
ドイツの研究チームは、タンパク質配列、三次元構造、機能特性に関するデータを統合し、メトリック学習に基づいてヒト E3 ユビキチンリガーゼの「パノラマビュー」を構築しました。
生物において、細胞タンパク質の適時な分解と再生は、タンパク質恒常性の維持に不可欠です。ユビキチン-プロテアソームシステム(UPS)は、シグナル伝達とタンパク質分解を制御する中核的なメカニズムです。このシステムにおいて、E3ユビキチンリガーゼは主要な触媒ユニットとして、特定の基質を認識し、ユビキチン標識を触媒することで、タンパク質の分解、局在、そして機能状態を制御します。さらに、E3リガーゼは免疫経路と炎症経路も制御します。E3リガーゼは組織特異的に発現し、発達症候群やメタボリックシンドローム(がんの進行を含む)との関連性から、特にこれまで創薬が困難であった標的に対する有望な創薬標的となっています。
E1酵素(約10種類)およびE2酵素(約50種類)と比較すると、ヒトE3リガーゼは多数(約600種類)同定されています。しかしながら、多くのヒトE3リガーゼは未だ部分的にしか特徴づけられておらず、仮説段階または未知のものも多数存在します。現在までに、研究された E3 リガーゼは高い異質性を示しています。このため、E3リガーゼは最も多様性に富んだ酵素クラスの一つとなり、パターン認識や大規模研究のボトルネックとなっています。したがって、ヒトE3リガーゼゲノム(ヒトゲノムにコードされているE3リガーゼの完全なセット)の詳細な特性評価と解析は、それらの生物学的機能を包括的に理解するために不可欠です。
この文脈では、ドイツのゲーテ大学の研究チームが「ヒトE3リゴーム」を分類した。タンパク質配列、ドメイン構成、三次元構造、機能、発現パターンなどの多段階データを統合します。チームの分類方法はメトリック学習パラダイムに基づいており、弱く監視された階層的フレームワークを採用して、E3 ファミリーとそのサブファミリー間の真の関係を捉えます。このアプローチは、E3 酵素の従来の分類 (RING、HECT、および RBR クラス) を拡張し、マルチサブユニット複合体とモノマー酵素を区別し、E3 酵素を基質および潜在的な薬物ターゲットにマッピングします。
「マルチスケール分類がヒトE3リゴームの複雑性を解読する」と題された関連研究結果が、Nature Communications誌に掲載されました。
研究のハイライト:
* 既存の E3 リガーゼのドメイン アーキテクチャ、3 次元構造、機能、基質ネットワーク、および小分子相互作用を分類フレームワークにマッピングして、一般的な洞察とファミリー固有の洞察を獲得します。
* 開発されたマルチスケール分類フレームワークは、典型的な E3 メカニズムと非典型的な E3 メカニズムの両方をカバーし、E3 リガーゼの広範な生物学的概要を理解するための完全なロードマップを提供します。
* E3 基質ネットワークに基づく薬物介入戦略の開発に新たな道を開きます。

用紙のアドレス:
https://www.nature.com/articles/s41467-025-67450-9
弊社の公式 WeChat アカウントをフォローし、バックグラウンドで「E3 酵素」と返信すると、完全な PDF を入手できます。
AIフロンティアに関するその他の論文:
https://hyper.ai/papers
データセット: ヒトE3ユビキチンリガーゼデータの構築
研究チームはまず、8 つの独立したデータ ソースからヒト E3 ユビキチン リガーゼ データを統合しました。過去の文献報告と公開データベース(E3Net、UbiHub、UbiNet 2.0、UniProt、BioGRIDなど)に基づき、1,448個のタンパク質エントリからなる予備データセットが作成されました。重複エントリおよび偽陽性の可能性のあるエントリは、様々なソースからのデータの相互比較と一貫性スコアリングによって除去されました。その後、InterProが提供するRING、HECT、およびRBR触媒ドメイン特性を用いて、462個の信頼性の高い触媒性E3ユビキチンリガーゼをスクリーニングし、最終的なヒトE3リガーゼゲノムを作成しました。
マルチサブユニットE3複合体(Cullin-RINGリガーゼなど)では、機能的に異なる3つのサブユニット(足場タンパク質、アプタマータンパク質、受容体タンパク質)が連携して、E2~Ub分子を特定の基質に局在させます。大きく、剛性があり、中央に位置する足場タンパク質(Cullinファミリー、Cul1~Cul5など)は、触媒RINGフィンガードメインサブユニットとアプタマー/受容体のドッキング部位に同時に結合することで、リガーゼ複合体全体を構成します。アプタマータンパク質はモジュールを橋渡しし、足場タンパク質のN末端ドッキング面を個々の基質受容体に接続します。受容体タンパク質は基質特異性を決定し、基質上の分解シグナル(デグロン)を直接認識して結合し、どの基質がユビキチン化されるかを決定します(例:Skp2、Keap1、VHL)。研究チームは、3 つのサブユニット(151 個のアプタマー、106 個の受容体、および 8 個のスキャフォールドタンパク質)を独立して注釈付けし、分類しました。彼らはまた、タンパク質間相互作用 (PPI) を使用して、マルチサブユニット E3 の基質をマッピングしました。
その後、触媒ドメインのスクリーニング段階で、研究者は触媒能力を主な基準として候補タンパク質を厳密に選別しました。システムは InterPro などのドメイン データベースを使用して、RING、HECT、RBR など、E3 活性に直接関連する主要な触媒ドメインを特定しました。これらのドメインを明示的に含み、配列レベルと構造レベルでユビキチン連結機能をサポートするタンパク質のみが、最終的な「触媒E3リガーゼ」の構築に保持されます。このプロセスにより、制御にのみ関与し、直接的な触媒能を持たない補助タンパク質が効果的に排除され、コアE3セットの機能的一貫性が確保されます。
メトリック学習に基づくマルチスケール分類フレームワーク
ヒトE3リガーゼゲノム内の複雑な関係を捉えるために、研究者たちは機械学習の手法を使って、新たな距離メトリックを学習しました。全体的なフレームワークは次の図に示されています。
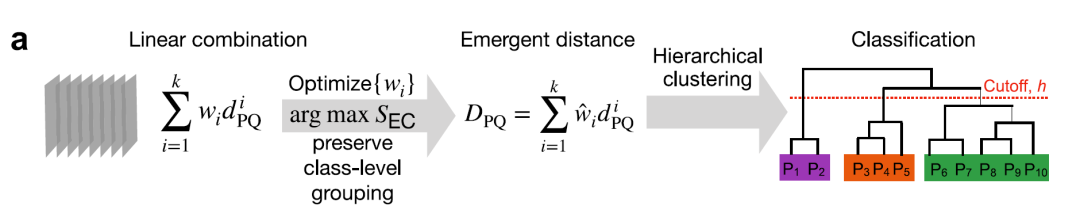
① マルチスケール距離測定
研究者らは、12 種類の異なる距離を計算することによって、E3 リガーゼ間のペアワイズ関係をエンコードしました。これらの距離は、一次配列、ドメイン構造、三次構造、機能、細胞内局在、細胞株/組織発現など、複数の細かいレベルをカバーします。下の図に示すように、すべての距離メトリックは比較と組み合わせのために [0,1] 間隔にスケーリングされます。
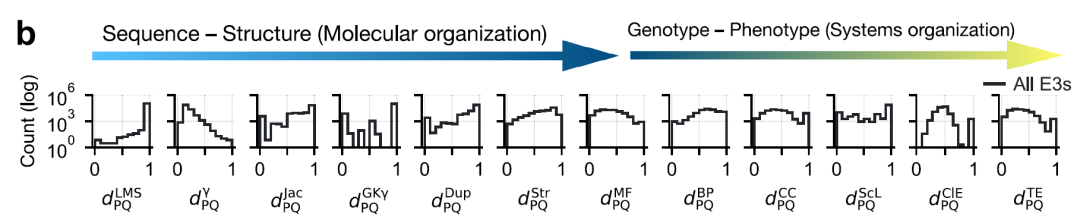
* シーケンスレベル: ローカル マッチング スコア (LMS) 距離 (ペアワイズ マッチングなし) とアライメント ベースの γ 距離が使用されました。
* ドメイン アーキテクチャ レベル: Jaccard 距離、Goodman-Kruskal γ 距離、ドメイン繰り返し距離の 3 つの距離が計算されました。
* 3D構造レベル: AlphaFold2モデルのTMスコアを使用
* 機能レベル: タンパク質と P および Q 間の機能距離は、GO アノテーションの意味的類似性を使用して測定され、次の 3 つのオントロジーをカバーします: * 分子機能 (MF)、生物学的プロセス (BP)、および細胞成分 (CC)。
* 細胞内局在距離
* 組織と細胞株間の共発現距離
②メトリック最適化、クラスタリング、ブートストラップ、分類
4 つの主要距離 (γ、Jaccard、構造、分子機能) に重み付けして統合し、下図に示すように、弱教師あり学習と要素中心類似度指標 (SEC) を通じて重みを最適化して、最適な複合指標を取得します。
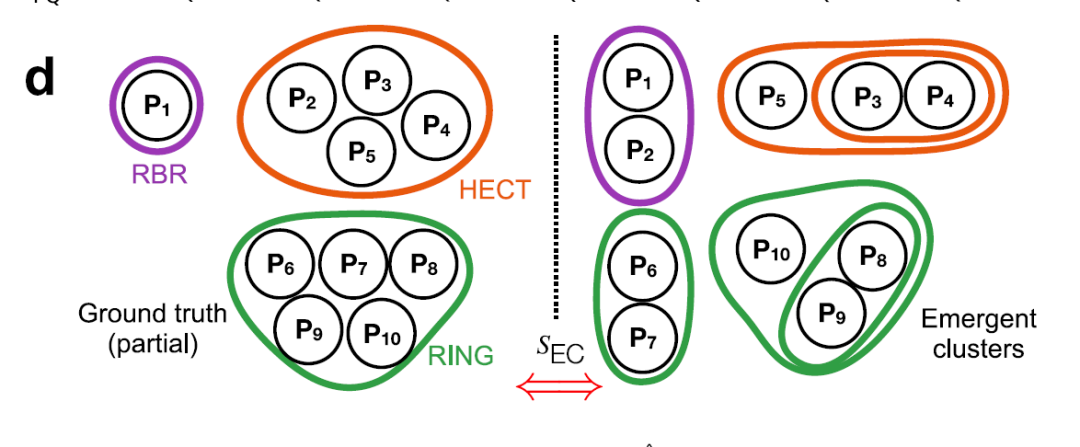
階層的クラスタリングは、Ward の最小分散法を使用して実行されました。最終的なE3デンドログラムを生成するために、ブートストラッピング法を用いて支持度が計算されます。最適な創発クラスターは、ツリースライス閾値h = 0.25を用いて得られ、462個のE3クラスターを13のファミリー(RINGファミリー10個、HECTファミリー2個、RBRファミリー1個)に体系的に分割します(下図参照)。
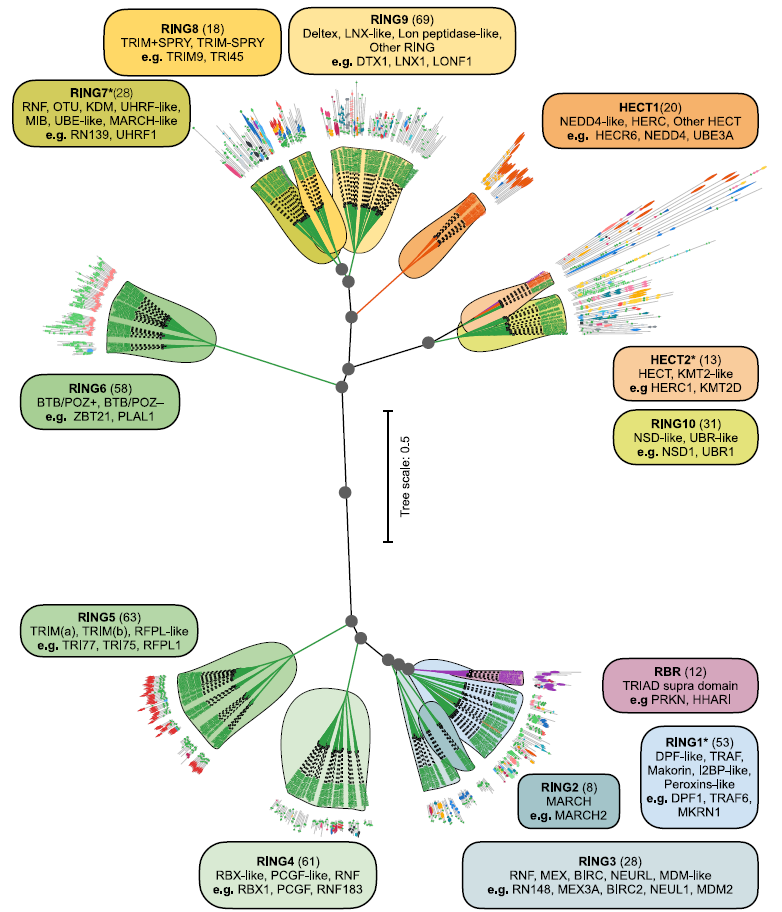
各ファミリーは、サブファミリーと異常なタンパク質を特定するために、配列とドメインの特徴についてさらに手動で分析されます。
③ 小分子のクラスタリングと結合確率
小分子クラスタリングのための統合2D UMAP投影局所的な密度ピークを組み合わせることで、20 個の代表的な小さな分子クラスターが特定されました。各クラスターが E3 タンパク質に結合する確率は、対数変換された傾向 (LPij) によって定量化され、その後の PROTAC 開発および標的小分子設計のガイドラインを提供します。
ヒト E3 リガーゼゲノムの完全性に関する詳細な評価が提供されました。
①ヒトE3リガーゼゲノムの詳細な構成
E3システムの体系化において、既存研究で用いられている多様な戦略と一貫性のない定義がもたらす課題に対処するため、本研究チームはE3システムの触媒成分を、1つ以上の触媒ドメインを含むポリペプチド配列として明確に定義しました。この客観的な基準により、E3の適切なアノテーションと標的解析が可能になります。最終的に研究者らは、データセット全体にわたる 462 個のポリペプチド配列に少なくとも 1 つの触媒ドメインが含まれていることを発見しました。これらのポリペプチドは、下の図に示すように、細かく組織化されたヒト E3 リガーゼゲノムを構成します。
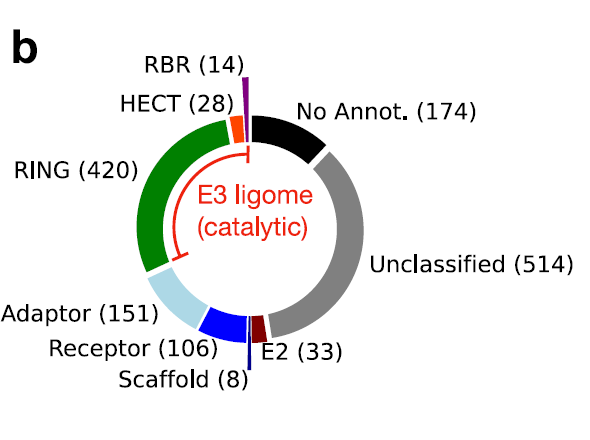
選別プロセスの信頼性を検証するために、研究者らは、さまざまなソースのデータセットにおける出現頻度に基づいて、各タンパク質のコンセンサススコアを定義しました。結果は、HECT および RBR クラス E3 リガーゼがデータセット内で非常に一貫していることを示しました (コンセンサス スコア ≥ 0.6、オレンジと紫のバー)。RING クラスのコンセンサス スコア (緑のバー) は広く分散しており、次の図に示すように、アノテーションの課題を示しています。
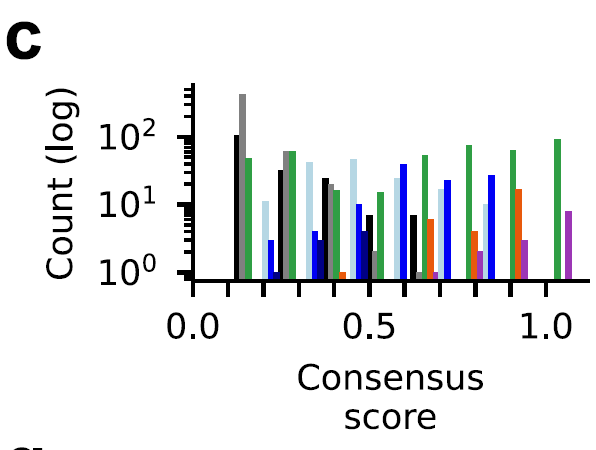
この方法を使用することで、研究者らは偽陽性と真陰性を最小限に抑え、信頼性の高い触媒活性E3を含め、疑似E3や触媒活性が検証されていない他のE3を考慮して、ヒトE3リガーゼゲノムの完全性の詳細な評価を行いました。
②ヒトE3リガーゼの機能分化
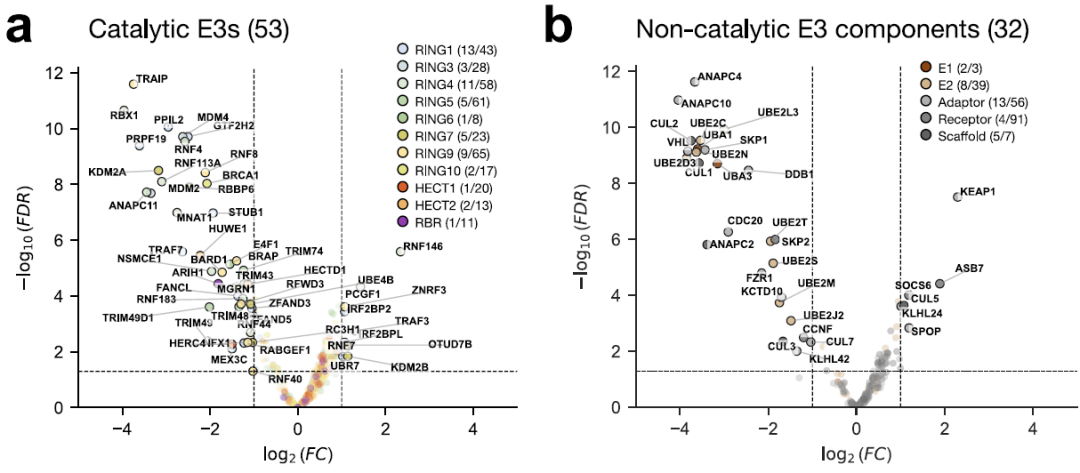
ヒトE3リガーゼの機能を評価するため、研究者らは細胞生存率を主要な表現型として、UPS遺伝子におけるCRISPR-Cas9欠失をスクリーニングした。その結果、…合計 53 個の触媒 E3 成分と 32 個の非触媒 E3 成分が、細胞の生存に不可欠であることが確認されました。以下に示すように:
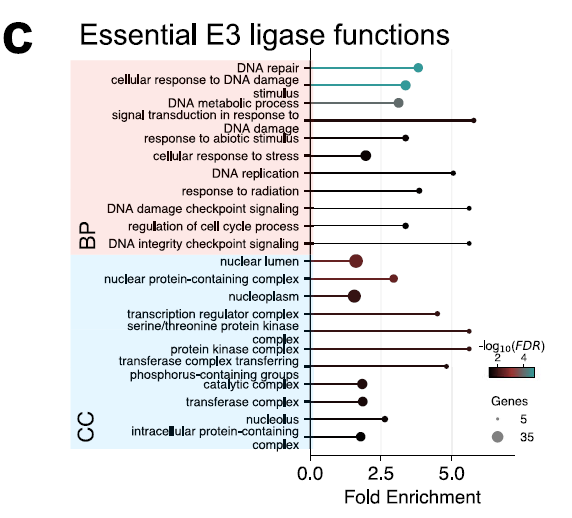
53種類の主要なE3のGO解析により、下図に示すように、E3は核内構成成分およびDNA損傷、複製、修復プロセスにおいて顕著に濃縮されていることが示され、ゲノムの完全性と核内制御の維持において中心的な役割を果たしていることが示唆されました。これらの結果は、細胞生存に不可欠なE3構成成分を明らかにしています。
Metascapeを用いて13のE3ファミリーに対してGOエンリッチメント解析を実施し、Cytoscapeを用いてネットワークを可視化しました。その結果、…異なるファミリーは、基質の選択、細胞の局在、および触媒機能において異なる役割を果たします。下の図に示すように、例えばRBRファミリーのメンバーであるRNF14、RNF144A、およびPRKNは、K6結合ユビキチンに特異的です。K6結合鎖は、停止したRNA-タンパク質架橋複合体(RNF14)、インターフェロンシグナル伝達を活性化するDNAセンシングアダプターSTING(RNF144A)、および除去のための損傷ミトコンドリア(PRKN)を標識することができます。同様に、TRIM E3 (RING5) は抗ウイルス自然免疫応答に著しく豊富に存在し、細胞内のパターン認識受容体の活動を制御します。RIG-1 および MDA5 によって媒介される反応など。
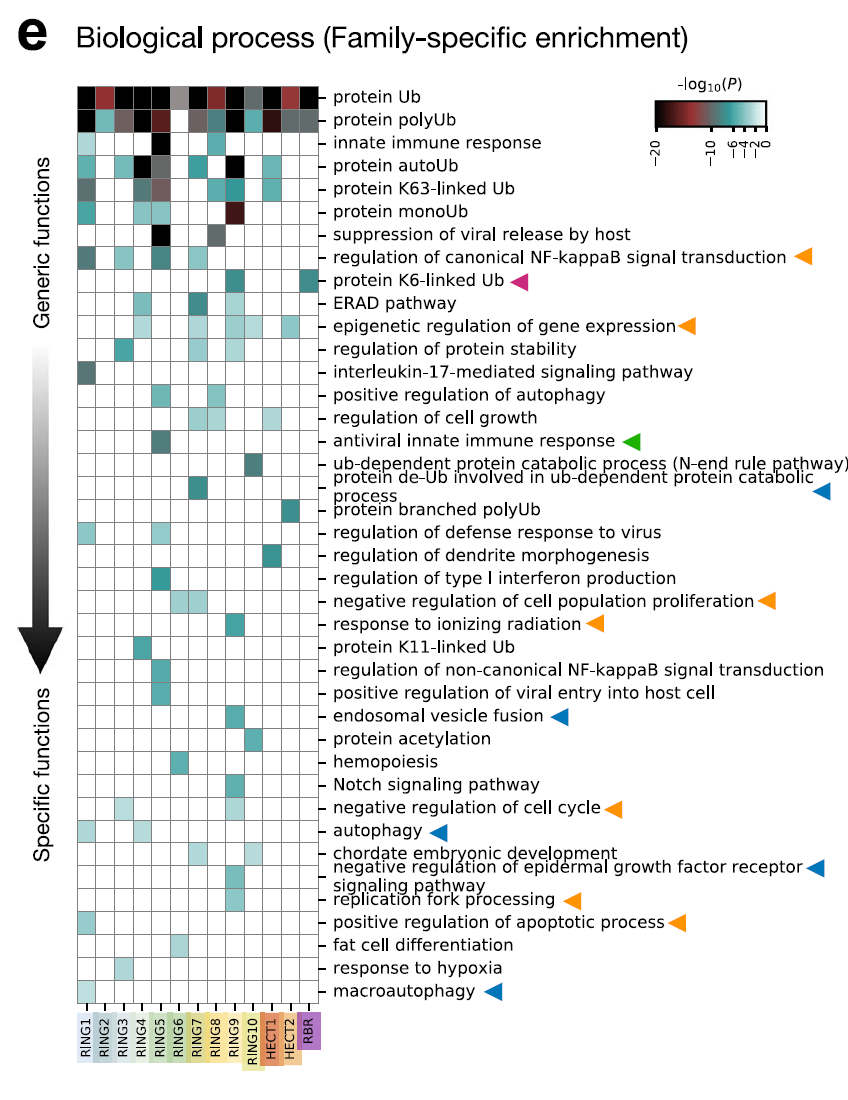
④ ヒトE3リガーゼのドラッグ化可能性マップ
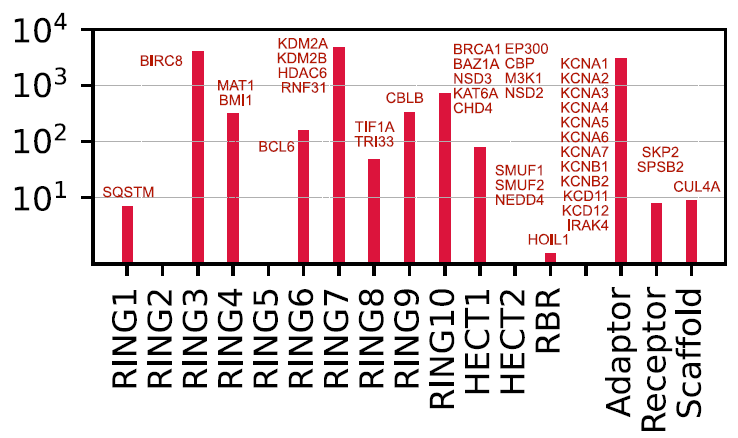
近位作用に基づく潜在的な治療経路を探索するため、研究者らは、既知のタンパク質分解標的キメラ(PROTAC)およびE3バインダー由来の既存のE3オペランドを、様々なE3およびそのファミリーにマッピングしました。現在、既存のE3オペランドが直接標的とすることができるタンパク質は16種類(9種類の触媒E3と7種類のアダプター)のみです。設計されたE3オペランドのほとんどはアダプタータンパク質(VHLやCRBNなど)を標的としますが、触媒E3(XIAP、MDM2/4/7、BIRC2/3/7など)を直接標的とするものはごくわずかです。
この研究でヒト E3 リガーゼを使用した最近傍分析により、5 つの高度に相関したタンパク質 (BIRC8、RN166/181/141、UBR2) が明らかになりました。以下に示すように:
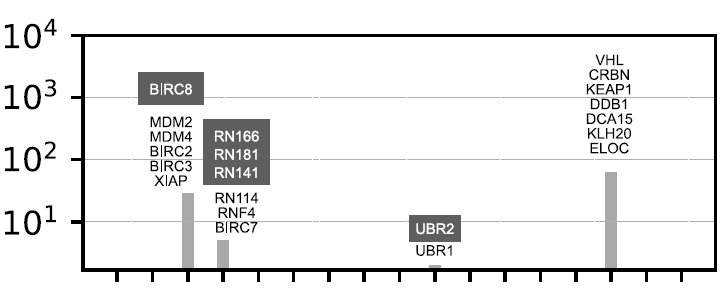
構造上の類似性が高いため (多くの場合、相同タンパク質)、既存の E3 オペランドを再利用してこれらのタンパク質をターゲットにすることができます。小分子 E3 バインダーのマッピングにより、研究者は追加の 25 個の E3 分子と 15 個の非触媒成分をターゲットにできる潜在的な化合物セットにアクセスできるようになりました。この発見により、未開発のターゲットが明らかになり、下の図に示すように、E3 オペランドのリード化合物を合理的に設計するための道筋が開かれました。
マルチスケール フレームワークは、複雑な生物システムを分析するための強力なツールを提供します。
機械学習分野において、マルチスケールフレームワークとは、異なる抽象度レベルや異なる特徴スケールでデータを処理できるモデリング手法または分析戦略を指します。これは固定されたアルゴリズムではなく、局所情報と大域情報、粗粒度と細粒度の特徴を統合し、モデルの表現力と汎化能力を向上させるための設計概念です。
マルチスケール分類フレームワークの価値は、E3リガーゼファミリー自体のシステマティックレビューに限定されません。より重要な意義は、オミクス統合のための移転可能かつ拡張可能なパラダイムを提供することにあります。このクロススケール統合アプローチは、他のマルチモーダルオミクスデータへの拡張を自然に可能にし、複雑な生物システムのシステマティック解析のための普遍的なツールを提供します。
例えば、細胞は生命の基本単位であり、その機能と運命は複雑な分子ネットワークによって決定されます。従来のディープラーニング手法は、単一細胞トランスクリプトームデータからの細胞種の識別においては優れた性能を発揮しますが、生物学的解釈可能性に欠けています。 2025 年 10 月 20 日、中国国家タンパク質科学センター (北京) と清華大学の研究者は、生物学的事前知識を統合するマルチスケールの解釈可能なディープラーニング フレームワークである Cell Decoder を提案しました。これにより、遺伝子や経路から生物学的プロセスまでの階層的な特性評価と推論が可能になり、単一細胞レベルで細胞の種類を解読する新しいアプローチが提供されます。 Cell Decoder は、タンパク質相互作用ネットワーク、遺伝子経路マッピング、経路階層関係をグラフ ニューラル ネットワーク アーキテクチャに埋め込むことで、クロススケールの生物学的知識グラフを構築します。研究チームは、公開されている7つの単一細胞データセットから得られたヒトおよびマウスのサンプルを用いて、Cell Decoderを9つの主流手法と比較し、体系的に評価しました。その結果、Cell Decoderは予測精度(0.87)とマクロF1(0.81)の両方で1位となり、ノイズ変動、細胞タイプの不均衡、バッチ間の分布シフトといった複雑な条件下でも安定した性能を維持しました。
論文のタイトル:Cell Decoder: マルチスケールの説明可能なディープラーニングによる細胞アイデンティティのデコード
用紙のアドレス:
https://link.springer.com/article/10.1186/s13059-025-03832-y
より長期的な視点から見ると、このマルチスケールフレームワークは、空間プロテオミクスデータ、低分子医薬品ライブラリー、化学空間情報とさらに統合することができ、基礎生物学研究、疾患メカニズム解析、トランスレーショナルアプリケーション間のデータ障壁を打破することが可能です。マルチオミクスデータの継続的な蓄積に伴い、このフレームワークはライフサイエンス研究とバイオメディカルイノベーションにおいてますます重要な支援的役割を果たすことが期待されます。
参考文献:
1.https://www.nature.com/articles/s41467-025-67450-9
2.https://blog.csdn.net/qazplm12_3/article/details/153948711
3.https://link.springer.com/article/10.1186/s13059-025-03832-y