Command Palette
Search for a command to run...
高度に選択的な基質設計を実現し、MIT とハーバード大学が協力して、生成 AI を使用して新しいプロテアーゼ切断パターンを発見します。

生体の複雑な生化学反応ネットワークにおいて、プロテアーゼはペプチド結合を特異的に切断することで、血液凝固や組織修復から免疫応答、さらには癌の進行に至るまで、一連の重要な生命プロセスを精密に制御しています。これらのプロテアーゼの機能不全は、しばしば様々な重篤な疾患の発生と進行に直接つながります。したがって、プロテアーゼの作用機序を解明し、その活性を精密に制御することは、基礎生命科学における中核的課題であるだけでなく、新たな診断法や治療法の開発に向けた重要なブレークスルーでもあります。
この目標を達成するための鍵は、鍵となるのは、高度に「適合」したペプチド基質を見つけることです。これらは、酵素の活性を追跡するための分子プローブとして使用したり、異常な活動をブロックするための阻害剤として設計したり、さらには標的治療を実現するための薬物送達システムで「条件付き活性化スイッチ」として機能することもできます。
しかし、標的プロテアーゼによって速やかに切断され、かつ高い選択性(その酵素によってのみ認識され、他のプロテアーゼとの交差反応を回避)を持つペプチド基質の設計は、科学界にとって常に大きな課題となってきました。この問題は、プロテアーゼと基質間の複雑な生化学的相互作用に起因しています。多様な生理機能に適応するために、プロテアーゼは広範な切断特異性を発達させており、その活性部位はペプチド基質(典型的にはアミノ酸約10個分)に正確に結合する必要があります。10個のアミノ酸からなる合成ペプチドのみを考慮した場合でも、一般的な天然アミノ酸20個を用いた場合、理論的な配列の組み合わせは約20¹⁰(約10¹³)に達し、探索の余地はほぼ無限に広がります。さらに問題を複雑にしているのは…同様の機能を持つプロテアーゼは、多くの場合共通の祖先に由来し、同様の活性部位構造を持つため、「相互認識」の影響を非常に受けやすくなります。このため、膨大な数の可能性の中から高度に特異的な基質を選別することが特に困難になります。
このボトルネックを克服するために、研究者たちは数々の試みを行ってきました。従来の方法は、天然タンパク質の既知の切断部位や酵素情報に頼ることが多く、効率が低く、理想的な人工基質を得ることが困難でした。化学生物学の知識に基づく合理的な設計は、通常、煩雑で、スループットが限られており、主に単一のプロテアーゼを対象としているため、スケールアップが困難です。近年、ハイスループットスクリーニング技術によって効率はある程度向上しましたが、操作の複雑さや高コストといった制約が依然として残っています。既存の計算予測方法のほとんどは、「切るかどうか」を決定することしかできず、切断効率を正確に分類することができないため、詳細なメカニズム研究やエンジニアリングアプリケーションのニーズを満たすことができません。
この文脈では、MIT とハーバード大学は共同で、人工知能に基づいたエンドツーエンドの設計フローである CleaveNet を提案しました。このアプローチは、予測モデルと生成モデルの相乗効果により、プロテアーゼ基質設計の既存のパラダイムに革命を起こし、関連する基礎研究と生物医学開発にまったく新しいソリューションを提供することを目指しています。

用紙のアドレス:
https://www.nature.com/articles/s41467-025-67226-1
弊社の公式 WeChat アカウントをフォローし、バックグラウンドで「CleaveNet」と返信すると、完全な PDF を入手できます。
AIフロンティアに関するその他の論文:
複数の実験シナリオのデータセットによってサポートされる、CleaveNet モデルの一般化機能を強化するクロスシナリオ検証。
本研究では、CleaveNet モデルの開発と検証において、モデルの信頼性と一般化能力を確保するために、配列構成と実験方法が大きく異なる 2 つのデータセットを統合しました。
研究者が使用したコアデータセットは、mRNAディスプレイ技術を使用して、18種類のマトリックスメタロプロテアーゼ(MMP)に対する約18,500種類の合成デカペプチドを含む基質ライブラリの切断活性を体系的に特徴付けた公開研究から得たものです。各基質-プロテアーゼの組み合わせは、相対的な切断強度を定量化するための標準化された切断効率スコア (Zₛₘ) に対応します。
評価の厳密性をさらに確保し、配列の類似性による過大評価を回避するために、研究者らは、最初のテスト セットに対して相同性フィルタリングを実行しました。研究者らは、各テスト配列とトレーニングセット内のすべての配列との間の最小レーベンスタイン距離を計算し、距離が3未満でトレーニングセットと非常に類似している816個の配列を除外した。最終的に、重複のない2,901個の配列を含む「mRNAディスプレイテストセット」が得られた。このサブセットはモデルトレーニングのどの段階でも使用されず、内部パフォーマンス検証のみに使用された。
大きく異なる生化学的背景に直面した場合のモデルの適応性を独立してテストするために、この研究では、「蛍光テスト セット」と呼ばれる完全に独立した分布外データセットも導入されました。このデータセットには、長さの異なる71種類の合成ペプチド(7~14アミノ酸)が含まれており、蛍光共鳴エネルギー移動(FRET)に基づく従来のin vitro実験を用いて、7種類の組換えMMPタンパク質に対する切断活性が検証されています。このデータセットは、ペプチド長の分布、アミノ酸組成、そして最も重要な実験的検出原理において、mRNAディスプレイ技術を用いて生成されたコアデータセットとは根本的に異なります。この意図的な設計は、CleaveNetモデルが特定の実験条件を超越し、普遍的な生化学パターンを捉える能力を評価するための重要なベンチマークとなります。
CleaveNet は、協調的な閉ループを予測して生成します。
下の図に示すように、CleaveNet の中核は、予測モジュール (CleaveNet Predictor) と生成モジュール (CleaveNet Generator) という 2 つの補完的かつ協調的な計算モジュールで構成されています。これらを組み合わせることで、完全な「設計評価」閉ループが形成されます。
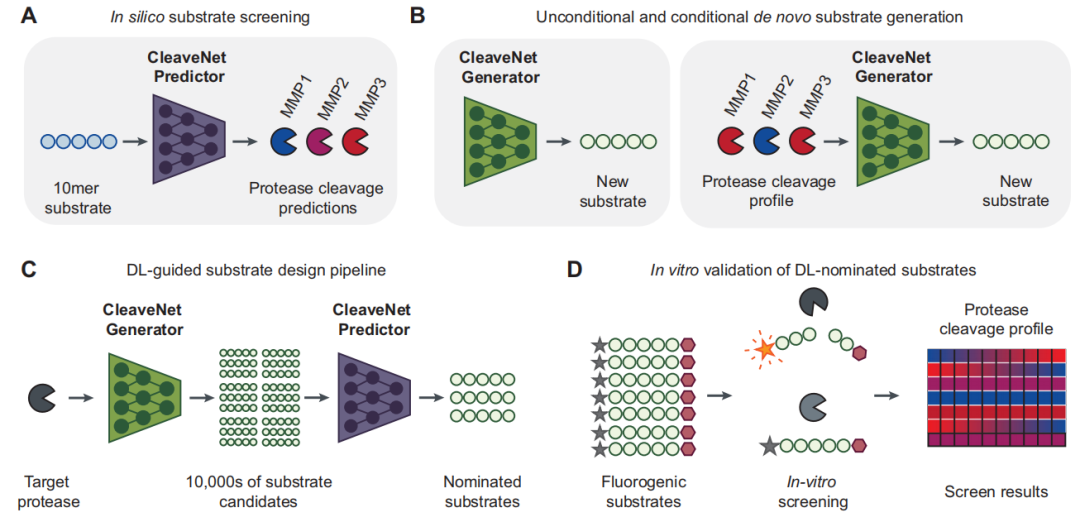
予測モジュールは、膨大な配列空間から候補基質の切断活性を迅速かつ正確に評価するという問題を解決することを目的としています。研究者らは、これを多出力配列関数回帰モデルとして構築しました。具体的には、このモデルはアミノ酸配列を入力として受け取り、その中心的なタスクは、18種類のMMPすべてについて、その配列の予測カットスコア(Ŵₛₘ)を同時に出力し、同時に各予測の不確実性(σₛₘ)を推定することです。
より高い予測の堅牢性を実現するために、本研究ではモデルアンサンブル戦略を採用しました。5つの同一の予測モデルをmRNAディスプレイトレーニングセットを用いて独立に学習させ、最終的な予測スコアはそれらの出力の平均とした。予測の不確実性は、これら5つの結果の標準偏差によって定量化された。さらに、調整可能な閾値(Zₜ)を設定することで、モデルは連続的な予測スコアを「切断」または「未切断」の2値判定に容易に変換し、異なるスクリーニングシナリオに対応できる。
本研究では、予測モデルの構築にあたり、シーケンスモデリングにおける2つの主流アーキテクチャ、すなわち双方向長短期記憶ネットワークとTransformerを体系的に比較しました。前者はシーケンスの依存関係を捉えることに優れており、後者はアテンション機構を備え、アミノ酸間の相互作用を包括的にモデル化できるため、現在、タンパク質言語表現の主流となっています。より大規模で多様なデータで実証されたその可能性に基づき、研究者は最終的に、CleaveNet Predictor の基盤として Transformer アーキテクチャを選択しました。
生成モジュールの目標は、候補基板の自動化されたインテリジェントな設計を実現することです。この研究では、自己回帰トランスフォーマーに基づく生成モデルをトレーニングし、mRNA 表現からデータセットに固有の普遍的な MMP 切断設定を学習できるようにしました。このモデルは、追加の入力条件なしで、多数の新規かつ合理的なペプチド配列を生成できます。
ランダム性を単純に再現するのではなく、生成モデルの価値を科学的に評価するために、研究者は「サイト非依存制御」と呼ばれる堅牢なベースライン手法を開発しました。この方法では、トレーニング データ内の各アミノ酸位置の独立した分布のみをカウントし、これに基づいてランダム サンプリングを実行してシーケンスを生成します。CleaveNet によって生成されたシーケンスをこのベースライン シーケンスと複数の次元にわたって比較することで、単純な統計的関連性を超えた、モデルによって学習された複雑な生化学的パターンを明確に識別できます。
予測モジュールと生成モジュールの緊密な連携により、研究者はまず多様な候補ライブラリを生成し、次にそれに対して効率的かつ正確な仮想スクリーニングを実行することができ、その結果、後続の実験検証のための強力な計算エンジンが提供されます。
CleaveNet は選択的かつ正確な制御を可能にします。
本研究では、モデル構築を完了した後、CleaveNetの性能について多段階かつ体系的な実験検証を実施し、その結果、予測精度、生成の合理性、実用化の有効性という点でこのプロセスの優れた価値が十分に実証されました。
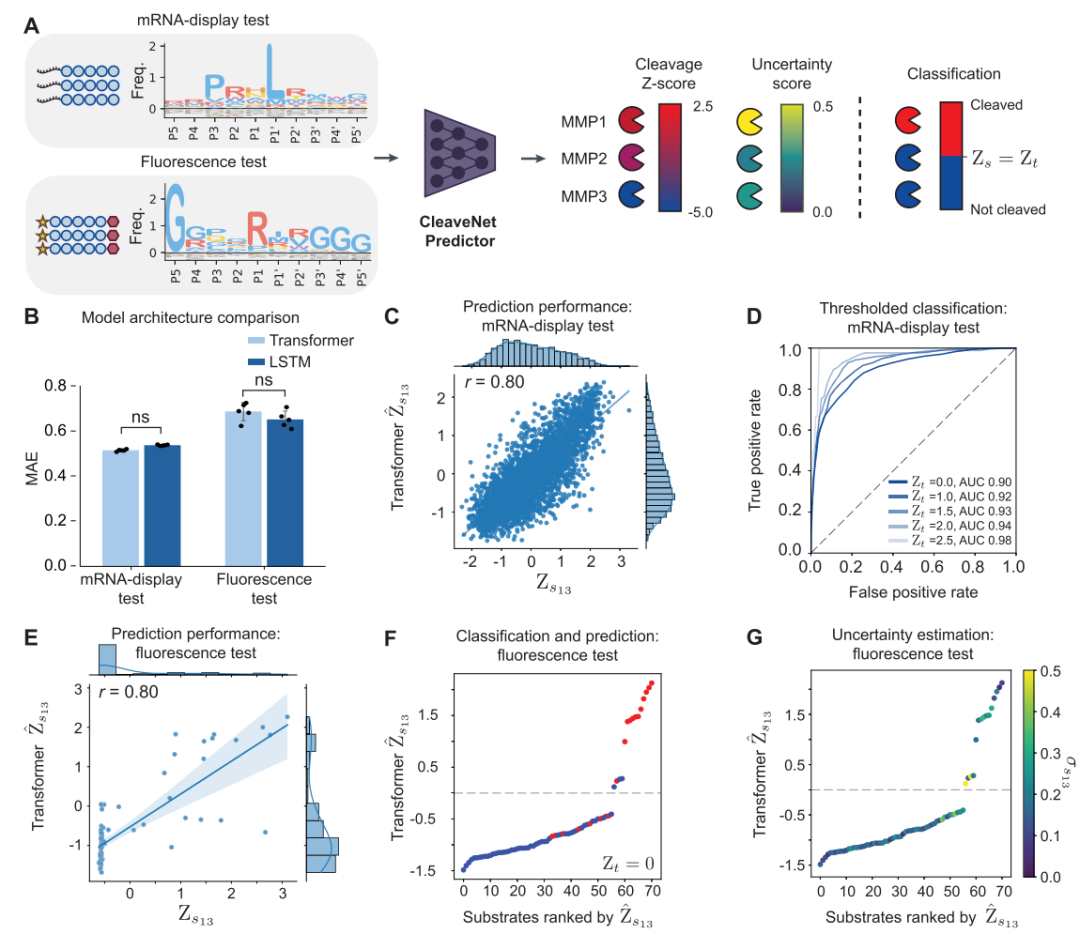
初め、CleaveNet Predictor は、内部テスト セットと外部テスト セットの両方で優れた予測機能を発揮します。訓練に一度も使用されなかった相同性フィルタリングテストセット(mRNAディスプレイテストセット)において、モデルのMMP13予測スコア(Ŵₛₘ)は、実験的に測定された標準化Zスコア(Zₛₘ)と強い相関を示しました(ピアソン相関係数r = 0.80)。連続予測を「カット/アンカット」の二値分類に変換した場合も、そのパフォーマンスは同様に堅牢でした。研究者らは、受信者動作特性(ROC)曲線をプロットし、曲線下面積(AUC)を計算することで、モデルが様々な決定閾値にわたって高い識別力を維持していることを発見しました。特に、一般的に受け入れられているカット閾値(Zₜ=2.5)では、AUCが0.98に達しました。より厳密なテストは、実験方法が大きく異なる、完全に独立した蛍光テストセットを用いて実施されました。
このデータセットの配列長、アミノ酸組成、検出原理はトレーニングデータと異なりますが、モデルの予測カットスコアは実験値と強い正の相関関係を維持しており(MMP13 の場合 r = 0.80)、実験的に検証された「カット」配列と「アンカット」配列を正確に区別できます。これは、CleaveNet Predictor がトレーニング データ パターンを記憶できるだけでなく、プロテアーゼによる基質切断を制御する普遍的な生化学法則も捉えられることを強く裏付けています。一般化能力が強いです。
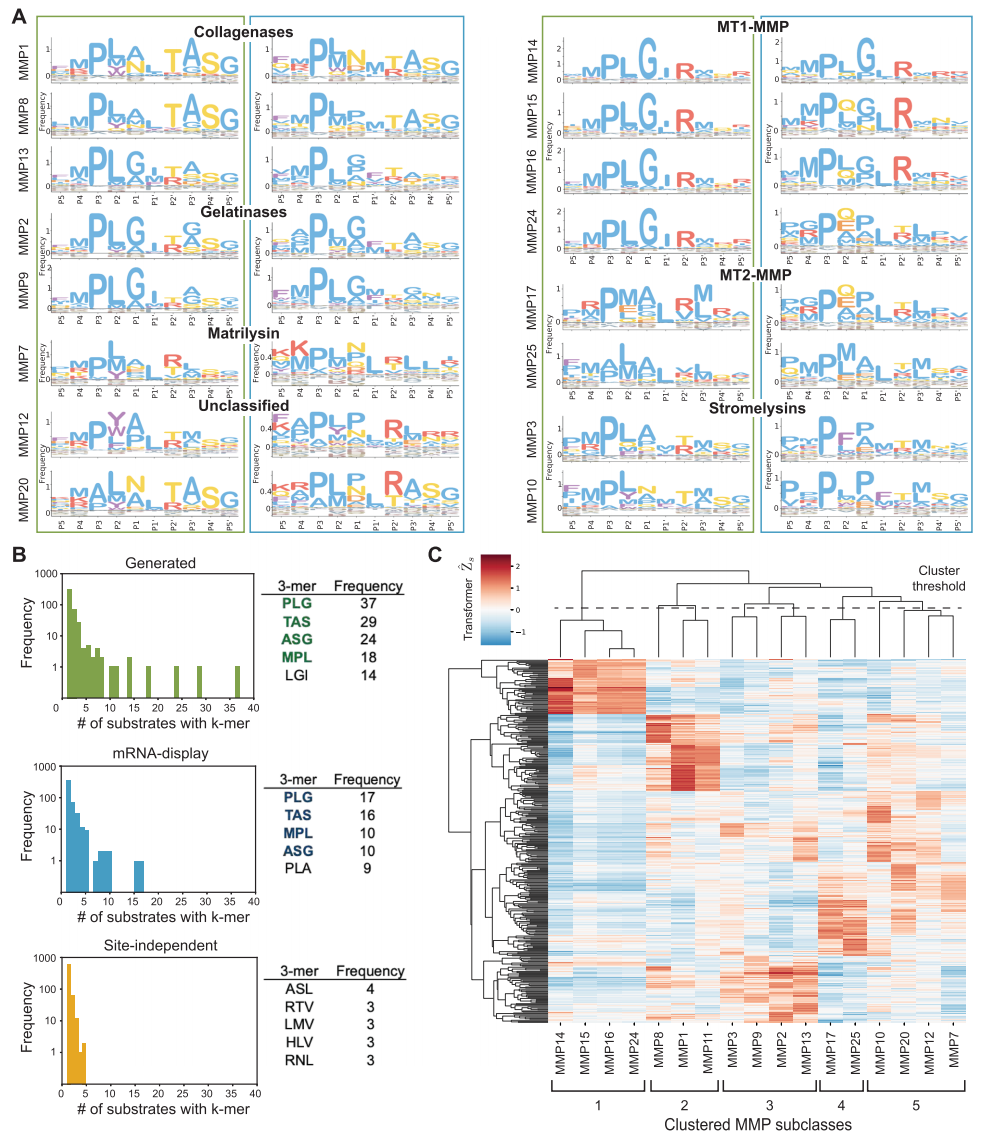
第二に、研究者による CleaveNet ジェネレーターによって生成された配列のバイオインフォマティクス分析により、その理論的根拠と新規性が明らかになりました。単一アミノ酸位置頻度のランダムサンプリングのみに基づく「部位非依存コントロール」配列と比較して、生成モデルによって生成された配列は、MMPファミリーの典型的な切断モチーフをより正確に再現し、主要な基質結合ポケット領域におけるアミノ酸分布が実際の実験データにより近いことを示しています。さらに重要なのは、生成された配列は、全体的な生体物理学的特性(疎水性や電荷など)の点で実際のデータセットと一致しています。しかし、高品質な生成とは、トレーニングデータの単純な複製を意味するものではありません。配列多様性解析の結果、生成された配列とトレーニングセットで共通するユニークな長鎖合成ペプチドの割合は極めて低いことが示されました。これは、モデルが過学習を回避し、トレーニングデータではカバーされていない新しい配列空間を探索できたことを示しています。
さらに機能クラスタリング解析を行ったところ、さまざまな MMP によって生成された高スコアの基質の予測切断活性スペクトルは、MMP 触媒ドメインの系統関係に基づいて自然にクラスタ化できることが明らかになりました。これは、生成モデルが見かけの配列パターンを学習するだけでなく、プロテアーゼの進化における機能的差異に関する情報も本質的に捕捉していることを示しています。これは、生成された結果の生物学的合理性を示しています。
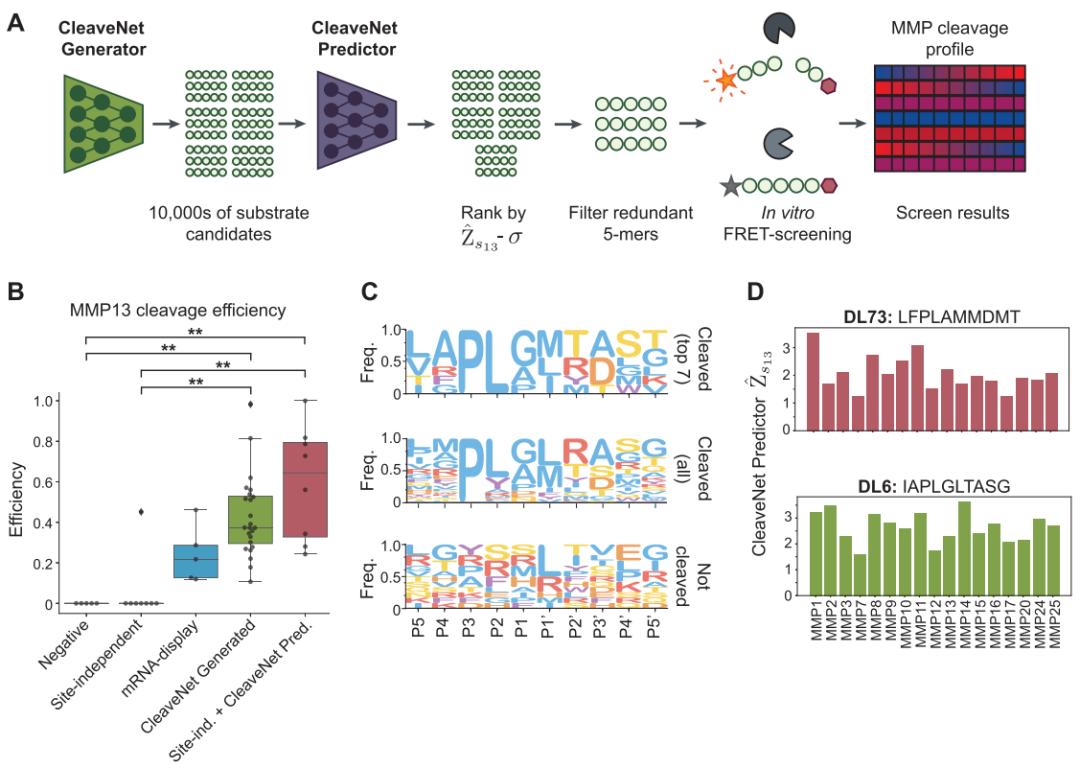
最終的に、すべての計算設計の妥当性はin vitro生化学実験によって検証されました。研究者らは、CleaveNetによって設計されたMMP13を標的とする候補基質の複数のセットを合成しました。これには、生成モデルによって直接生成された配列と予測モデルによってスクリーニングされた配列が含まれます。蛍光共鳴エネルギー移動(FRET)切断実験では、説得力のある結果が得られました。CleaveNet パイプラインを使用して設計された 24 個の基質はすべて、再構築された MMP13 によって正常に切断され、ヒット率 100% (TP3T) を達成しました。さらに、切断効率の中央値は、トレーニングセットに含まれる既知の高効率ポジティブコントロール基質よりも有意に高かった。これは、このプロセスが高効率基質を設計する能力を実証している。
本研究では、高選択性基質の設計といったより困難な課題への取り組みにおけるこのプロセスの可能性を実証するため、条件付き生成戦略をさらに採用し、生成モデルの目的として「MMP13に対する高い選択性」を指定した。その後の大規模並列in vitroスクリーニング(12種類のMMPについて95の基質ペア)により、条件付きガイダンスによって生成された基質は高い選択性を示すことが示された。切断活性は MMP13 に大きく偏っており、選択性が高くなります。
特に注目すべきは、設計された基質の一部が、高い切断効率と高い選択性の両方を備えていることです。これは、元のトレーニング データでは非常にまれな優れた組み合わせであり、CleaveNet が新しい高品質のシーケンス空間を探索する強力な能力を際立たせています。
要約すると、正確な計算予測から合理的な配列生成、そして最終的なウェット実験検証に至るまで、一連の相互に関連した結果は、CleaveNetが効率的で信頼性が高く、強力なプロテアーゼ基質設計プラットフォームを構築したことを示しています。この研究は、プロテアーゼ活性制御という古典的な課題に対する革新的なAIソリューションを提供するだけでなく、将来のプロテアーゼ機能研究と関連医薬品開発のための新たな方法論的基盤を築くものでもあります。
プロテアーゼ基質設計におけるAI主導のイノベーション
CleaveNet の AI 支援プロテアーゼ基質設計テクノロジーは、世界中のライフサイエンスおよびバイオメディカル分野におけるイノベーションを推進しています。
ワシントン大学のデビッド・ベイカー氏のチームが、画期的な研究をサイエンス誌に発表した。初めて AI を活用して、複雑な活性部位を持つセリン加水分解酵素をゼロから設計しました。これは、知られている酵素ファミリーの中で最大のものの 1 つです。この研究では、新しい機械学習ネットワークであるPLACERが導入され、エステル加水分解を効率的に触媒できる活性酵素の設計に成功しただけでなく、予想外に5つの新しいタンパク質折りたたみパターンを発見し、この酵素ファミリーの構造的多様性を大幅に拡大しました。
* 論文タイトル: セリン加水分解酵素の計算設計
* 論文リンク:
https://www.science.org/doi/10.1126/science.adu2454
さらに、ヨーロッパの複数の大学による共同チームが、Transformerアーキテクチャを基盤とした、プロテアーゼと基質の相互作用を正確に予測できる汎用モデルを開発しました。このモデルは、複数のソースから収集されたプロテアーゼの切断データを統合し、種を超えた基質配列の効率的な予測を実現します。その汎用性は、細菌やウイルスを含む様々な病原体のプロテアーゼに関する研究で検証されており、抗感染症薬開発における重要な配列設計基盤を提供しています。
計算生物学、人工知能、そして合成生物学の融合が進むにつれ、プロテアーゼ基質設計は、芸術と経験を融合させた科学から、高度に合理化され工学的に設計された研究分野へと進化することが予測されます。これは、新薬、診断ツール、そしてグリーンバイオ触媒の開発を加速させるだけでなく、最終的には生命制御の根底にあるロジックを解読し、生命機能をオンデマンドでプログラミングする新しい時代を切り開く可能性を秘めています。








