Command Palette
Search for a command to run...
無秩序なタンパク質アセンブリの予測力を再構築するために、NVIDIA、MIT、オックスフォード大学、コペンハーゲン大学、ペプトンなどが生成モデルと新しいベンチマークをリリースしています。

構造生物学の歴史において、「構造が機能を決定する」という原理は、かつてはほぼ揺るぎない基本法則と考えられていました。インスリンの典型的ならせん構造とヘモグロビンの四量体構造は、タンパク質が生物学的機能を発揮するためには安定した三次元構造を持たなければならないというコンセンサスを強固なものにしました。
しかし、本質的に無秩序なタンパク質(IDP)とその本質的に無秩序な領域(IDR)の発見この伝統的な理解は絶えず変化し続けています。これらのタンパク質は生理学的条件下では固定された構造を形成することはありませんが、シグナル伝達や遺伝子転写制御といった中核プロセスに深く関与しており、がんや神経変性疾患といった主要なヒト疾患とも密接に関連しています。
計算生物学研究により、真核生物のプロテオーム中の約301個のTP3Tアミノ酸残基が無秩序状態にあることが明らかになりました。これは、無秩序は「異常」ではなく、むしろ生体システムの正常な構成要素であることを意味します。しかしながら、無秩序なタンパク質は極めて動的な性質を持つため、従来の実験技術を使用して安定的に捉えることが困難であり、従来の計算方法を使用してその構造分布を正確にシミュレートすることも困難です。これは、この分野における長年の技術的なボトルネックとなっています。
この課題に対処するため、英国を拠点とするタンパク質分析技術開発企業であるペプトン、コペンハーゲン大学、NVIDIA、オックスフォード大学、MIT、デューク大学などからなる共同チームが、2つの重要なブレークスルーを提案しました。1つは、PeptoneBench システム評価フレームワークです。このフレームワークは、SAXS、NMR、RDC、PRE などの複数のソースの実験データを統合し、最大エントロピー再加重などの統計手法を組み合わせて、実験観察と理論予測の厳密な定量的比較を実現します。2番目は生成モデルPepTronです。拡張された合成 IDR データセットのトレーニングにより、無秩序領域をモデル化する能力が特に強化され、無秩序タンパク質の構造多様性をより適切に捉えることができるようになります。
研究チームはPeptoneBenchを用いて、PepTronをAlphaFold2、Boltz2、BioEmuといった主流の予測ツールと体系的に比較しました。その結果、PepTronは秩序領域と無秩序領域の両方の予測において実験結果と高い整合性を示し、最先端(SOTA)の性能に達していることが示されました。これらの進歩に基づき、「コンフォメーションセット」を用いてタンパク質構造を予測するための、より正確で生物学的に現実的なフレームワークが出現しつつあり、タンパク質の秩序・無秩序スペクトル全体にわたる理解を大幅に向上させます。
「秩序-無秩序連続体全体にわたるタンパク質アンサンブル予測の進歩」と題された関連研究成果が、bioRxiv にプレプリントとして公開されました。

用紙のアドレス:
https://www.biorxiv.org/content/10.1101/2025.10.18.680935v1
弊社の公式 WeChat アカウントをフォローし、バックグラウンドで「PepTron」と返信すると、完全な PDF を入手できます。
AIフロンティアに関するその他の論文:
https://hyper.ai/papers
PeptoneBenchとマルチソース実験データセットの体系的な構築
タンパク質データベース (PDB) は構造生物学における最も基本的かつ重要な公開リソースですが、本質的に無秩序なタンパク質 (IDP) とその無秩序領域 (IDR) のカバー範囲には大きな構造的ギャップがあります。約 3% エントリのみが順序なしとしてマークされました。しかし、ヒトのプロテオームでは、このような無秩序な領域の割合は 20~30% と高くなります。
下の図に示すように、この体系的なバイアスにより、ほとんどの構造予測モデルは自然に安定なコンフォメーションを「好む」ようになり、長期的には動的で無秩序な状態から学習する能力が制限されます。この欠陥を補うために、研究者らは、約 771 TP3T の順序付けられていない割合を持つ IDRome などの補足データベースを導入しました。統計分布の面ではPDBを補完することができます。しかし、このデータベースには実際の実験で解析された構造データが不足しており、モデリングや評価の直接的なベンチマークとして使用することは困難であり、その応用価値は依然として著しく限られています。
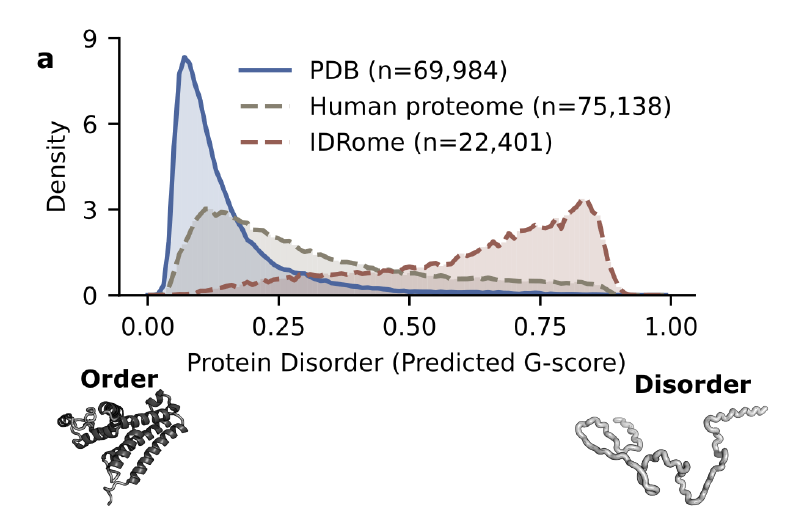
前述のデータのボトルネックを克服するために最初のステップは、障害の定量化および比較が可能な指標を確立することです。本研究では、タンパク質の平均Gスコアを中核指標として用い、その値は0(完全に秩序化)から1(完全に無秩序化)までの範囲で表されます。NMR二次化学シフト(CS)データに基づいて算出されたこのスコアは、局所的な二次構造形成の傾向を正確に反映しています。実験的なCSデータが存在しないタンパク質については、研究チームはTriZODでトレーニングされたADOPT2機械学習モデルを用いてGスコアを予測し、秩序・無秩序スペクトル全体の統一的な定量化を実現しました。
これを踏まえて、研究チームはさらに、PDB の構造データだけに頼っていては、立体配座セットの品質を客観的に評価することはできないと指摘しました。したがって、順序付きと順序なしの範囲全体をカバーする実験データセットを構築する必要があります。
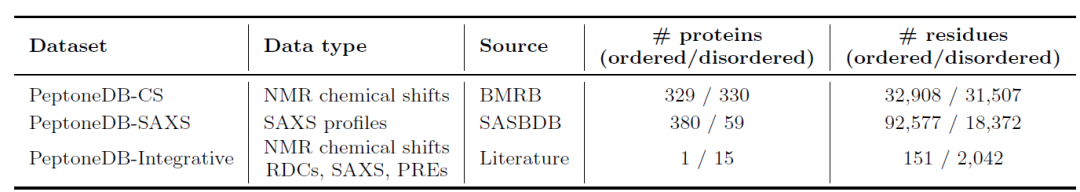
この目的のため、研究者らは下表に示すように、3つの補完的なデータリソースを構築しました。PeptoneDB-CS(BMRB由来のNMR化学シフト)、PeptoneDB-SAXS(SASBDB由来のSAXSスペクトル)、そしてPeptoneDB-Integrative(複数の直交実験データを統合した専用のIDPセット)です。これら3種類のデータはそれぞれ異なる構造を持ち、互いに補完的な情報を持っています。CSは局所的な構造を明らかにし、SAXSは全体的なコンフォメーションを反映し、Integrativeはクロスバリデーションをサポートします。
このデータに基づくと、下の図のようになります。研究者らは、予測されたコンフォメーション セットと実験データ間の一貫性を定量化するために、PeptoneBench 評価フレームワークを開発しました。全体のプロセスには、コンフォメーションセットの標準化と前処理、順モデルを用いた予測構造と実験と同等の観測値のマッピング、そしてプロセス全体を通してモデルと実験の両方からの不確実性を考慮した、正規化RMSEに基づく一貫性スコアリングが含まれます。最終結果はRMSE-Gスコアグラフとして提示され、Lowesスムージングとブートストラッピングを用いて誤差が推定され、さらにPeptoneBench集計スコアに統合されます。これにより、異なるツールの性能を直接比較するための定量的な基準が形成されます。
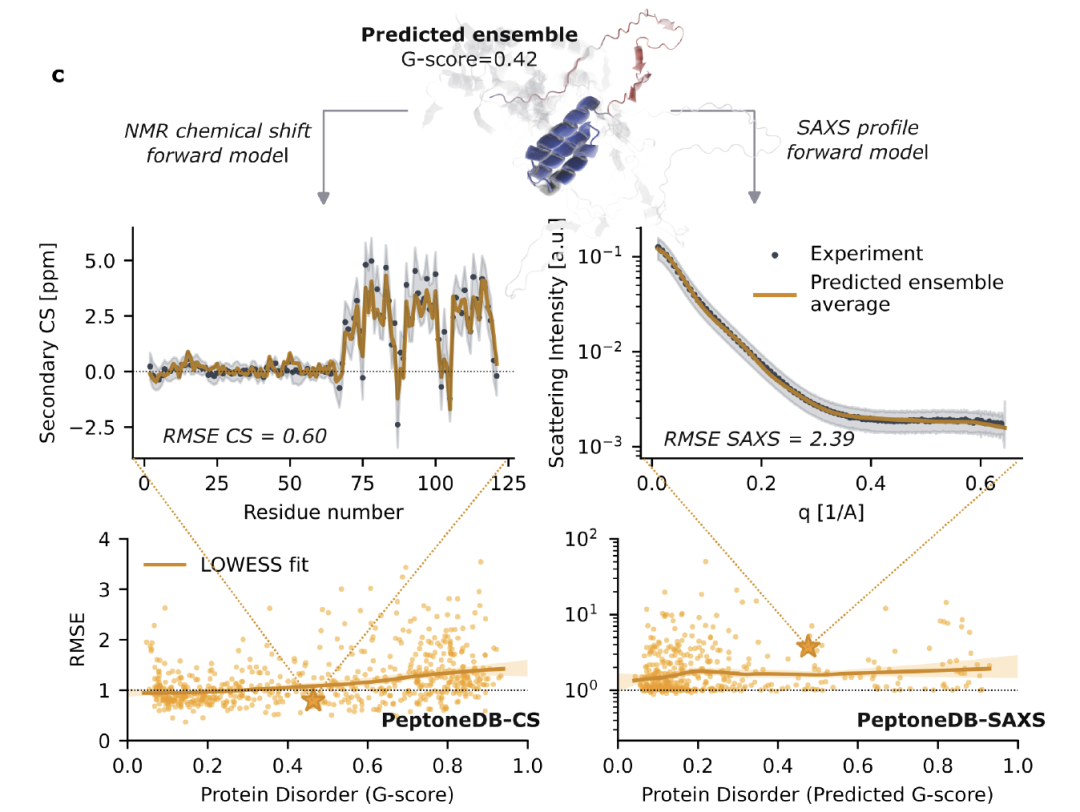
高いRMSEを示した初期のコンフォメーションセットの中には、最大エントロピーを用いて重み付けし直すことで、実際には実験分布に近づく可能性がある点を強調しておく価値がある。「誤った重み」を「欠落したコンフォメーション」と誤認することを避けるため、PeptoneBench は、修正可能なサンプリング バイアスと回復不可能な構造損失を区別するために、再加重の前後の RMSE も報告します。この戦略は、非常に動的で実験条件に極めて敏感な IDP にとって特に重要です。生成モデルが十分に豊富なコンフォメーション空間をカバーできる限り、実験環境が異なっていても再重み付けプロセスを通じて迅速に適応できるため、予測結果の実用性と信頼性が大幅に向上します。
ペプトロン:秩序だったタンパク質と無秩序なタンパク質のバランスをとる構造モデル
提案されたPepTronモデルは、ESMFlowフローマッチングアーキテクチャ上に構築されたタンパク質コンフォメーションジェネレータです。その目標は、完全に秩序だった構造から高度に無秩序な構造まで、あらゆるコンフォメーションスペクトルを網羅し、物理的に妥当でありながら構造的に多様なコンフォメーションセットを生成することです。
モデルアーキテクチャの観点から言えば、PepTron は ESMFlow に基づいており、トレーニングと推論の効率を向上させるために NVIDIA BioNeMo フレームワークに実装されています。このモデルはcuEquivariance三角アテンション機構を統合し、BioNeMoのModular Co-Designサブパッケージを通じてフローマッチング機能をサポートしています。学習プロセスはBioNeMoの分散ベストプラクティスに準拠しており、複数の並列戦略と混合精度計算を組み合わせることで、マルチGPU環境における安定的かつ効率的なスケーリングを実現します。
PepTronは推論フェーズにおいて多重配列アライメント(MSA)や外部ESM重みに依存しないことは特筆に値します。PepTronは単一のチェックポイントのみで完全なコンフォメーションセットを生成できるため、使用開始時の閾値が大幅に簡素化されます。
不規則領域における実験的構造データが不足しているという課題に対処するため、研究チームはIDRomeをベースとした合成構造データセットIDRome-oを構築した。そのため、彼らは、物理的に妥当な IDP 立体配座のセットを大規模かつ極めて低コストで生成できる、フラグメントアセンブリベースのタンパク質構造生成ツールである IDP-o を開発しました。 IDP-o はフラグメントアセンブリと階層的鎖成長戦略を組み合わせて、2 億 1,400 万の構造を含む AlphaFold データベースから 6 残基のフラグメントを抽出し、無秩序なタンパク質内の一時的ならせん構造をより正確に捉えます。
IDR-oの目的は、特定の平衡分布をシミュレートすることではなく、配列がサンプリングする可能性のあるすべての合理的なコンフォメーションをカバーすることであることに留意してください。したがって、その出力は、後続の最大エントロピー再重み付けに特に適しており、分子動力学シミュレーションのための高品質な初期コンフォメーションライブラリとしても利用できます。
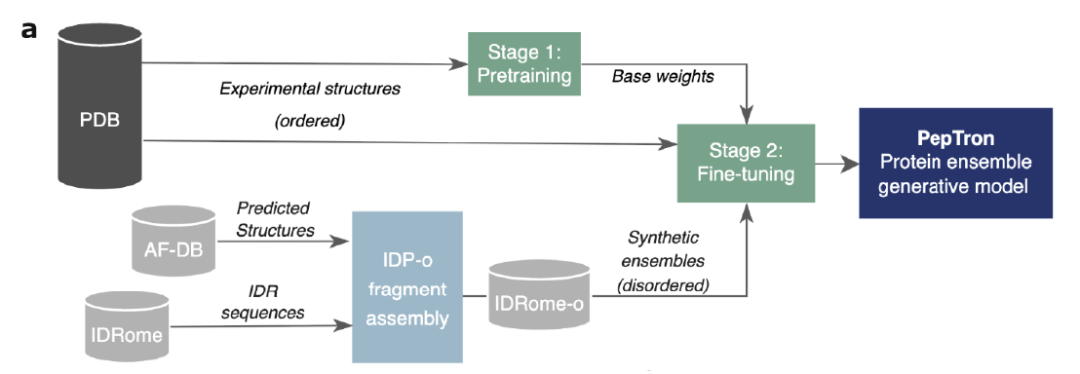
下図に示すように、安定した構造を予測する傾向がある従来のモデルのバイアスを克服するために、PepTron は、実験データと合成データを組み合わせたハイブリッド トレーニング戦略を採用しています。まず、PDBデータベースから実験的に解明された構造を用いてモデルを事前学習します。次に、合成的に生成された無秩序タンパク質のセットを導入して微調整を行い、モデルが秩序構造と無秩序構造の連続分布を完全に学習できるようにします。計算量が制限された条件下でも、この戦略により、様々なタンパク質に対するモデルの予測性能が大幅に向上します。
具体的な訓練手順としては、研究は2つの段階に分かれています。初期段階では、ESMFoldの重み付けから開始し、フローマッチングモジュールをPDBデータを用いて再学習させ、配列長トリミング範囲を512残基まで拡張します。ハイブリッド微調整段階では、PDB実験構造とIDRome-o合成データからなるハイブリッドデータセットを学習データとして用いて、モデルの最終的な最適化を行います。この設計により、PepTronは秩序-無秩序スペクトル全体にアクセスでき、タンパク質の動的コンフォメーション空間のより包括的かつ現実的なモデリングを実現します。
フルスペクトルコンフォメーションのモデル検証:ペプトロン法と主流法の体系的比較
研究チームは次に、PeptoneBenchフレームワークを用いて、トレーニングセットとは完全に独立した実験データにおけるPepTronの性能を体系的に評価し、ESMFold、ESMFlow、AlphaFold2、Boltz2、BioEmuといった主流のモデルと比較しました。同時に、チームは天然変性タンパク質(IDP)に焦点を当てたPeptoneDB-Integrativeデータセットで具体的なテストを実施し、各モデルの無秩序構造モデリング能力を包括的に検証しました。その結果、モデル間に明確な差別化特性が見られました。
下の図に示すように、PeptoneDB-CS データセットでは、各モデルのパフォーマンスはタンパク質の無秩序度 (G スコア) によって大きく異なります。ESMFold と ESMFlow は秩序だった領域の予測では正確ですが、無秩序な領域ではパフォーマンスが大幅に低下します。IDP-o は典型的な補完パターンを示し、無秩序度が高いほどパフォーマンスが向上します。PepTron は、秩序-無秩序構造スペクトル全体にわたって安定した高い一貫性を維持します。このバランス調整能力は、PeptoneDB-SAXS データセットとその後の再加重分析でさらに検証され、PepTron は秩序だった構造の精度を犠牲にすることなく、無秩序なタンパク質の構造多様性を効果的に捉えることができることが実証されました。
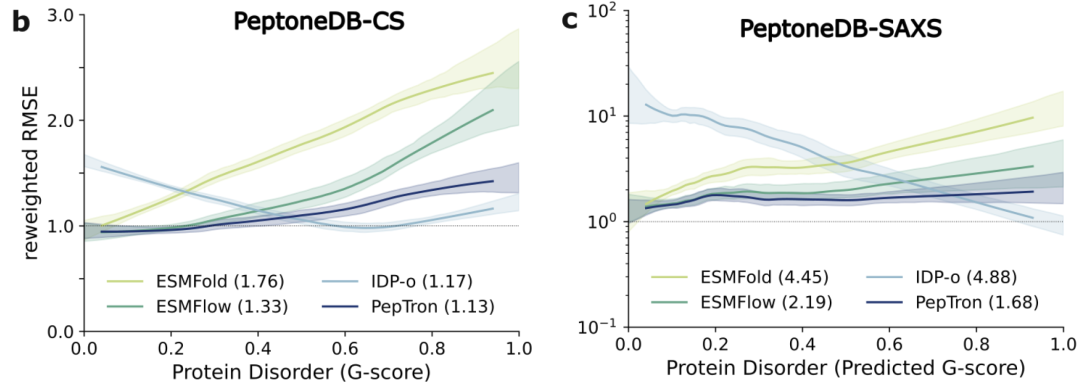
さらなるモデル間比較の結果は下図に示されています。AlphaFold2とBoltz2は依然として秩序あるタンパク質の予測において優位に立っていますが、無秩序度が増すにつれてその性能は系統的に低下します。対照的に、PepTron と BioEmu は、コンフォメーション スペクトル全体にわたって強力な堅牢性を示し、IDP の非常に異質な構造特性の処理により適しています。
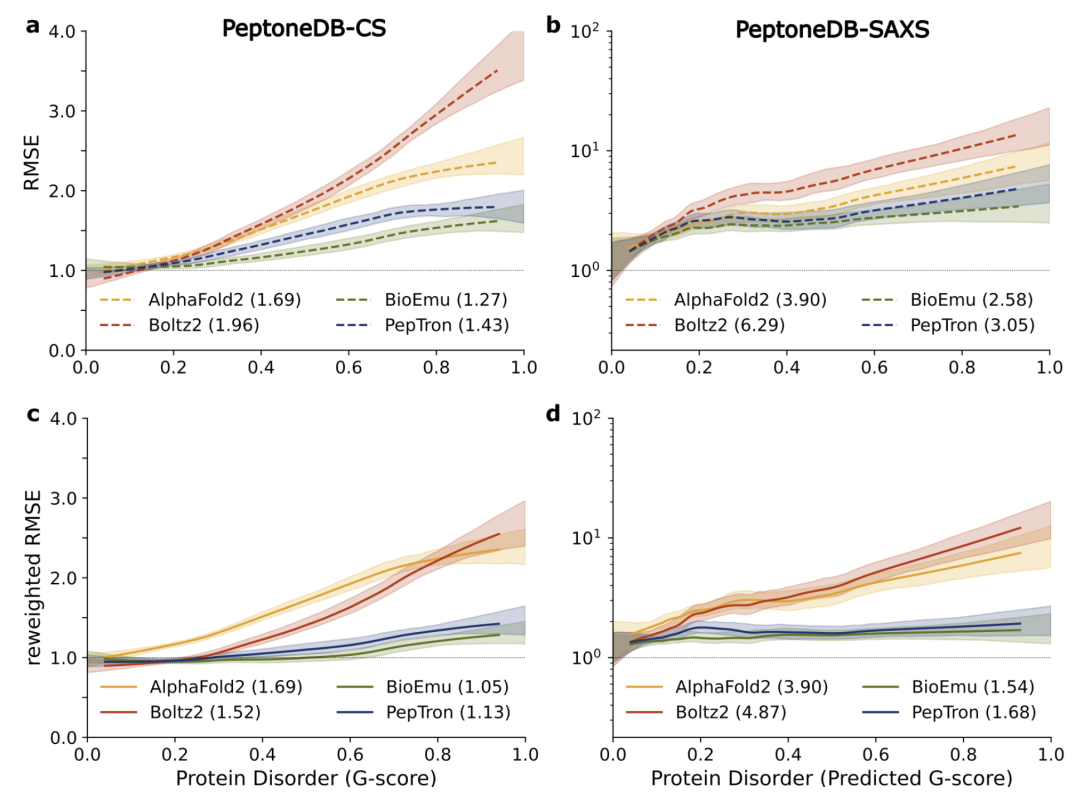
不規則領域での学習が秩序だったタンパク質の予測能力を損なわないことを確認するため、研究チームはCAMEO22とCASP14の秩序だった構造データを用いた追加テストも実施しました。その結果、…PepTron は、RMSD、LDDT、TM などの主要なメトリックに関して ESMFlow と同等のパフォーマンスを発揮し、IDR モデリング機能を拡張しながらも秩序だった構造の精度を損なわないことを実証しています。
下図に示すように、複数の実験指標を統合したPeptoneDB-Integrativeデータセットでは、モデルのパフォーマンスにさらなる違いが見られます。IDP-oは最大エントロピーの再重み付け後に特に優れたパフォーマンスを示し、RMSEとRDC Q値の両方で他のモデルを大幅に上回りました。PepTronとBioEmuはRDC指標では同等ですが、BioEmuは局所的な化学シフトの予測においてより有利です。重み付けされていない条件でも、以下の点が注目に値します。IDP-o は、ほとんどのローカルおよびグローバル メトリックで引き続きリードしており、無秩序なタンパク質構造をカバーする上での本来の利点を示しています。
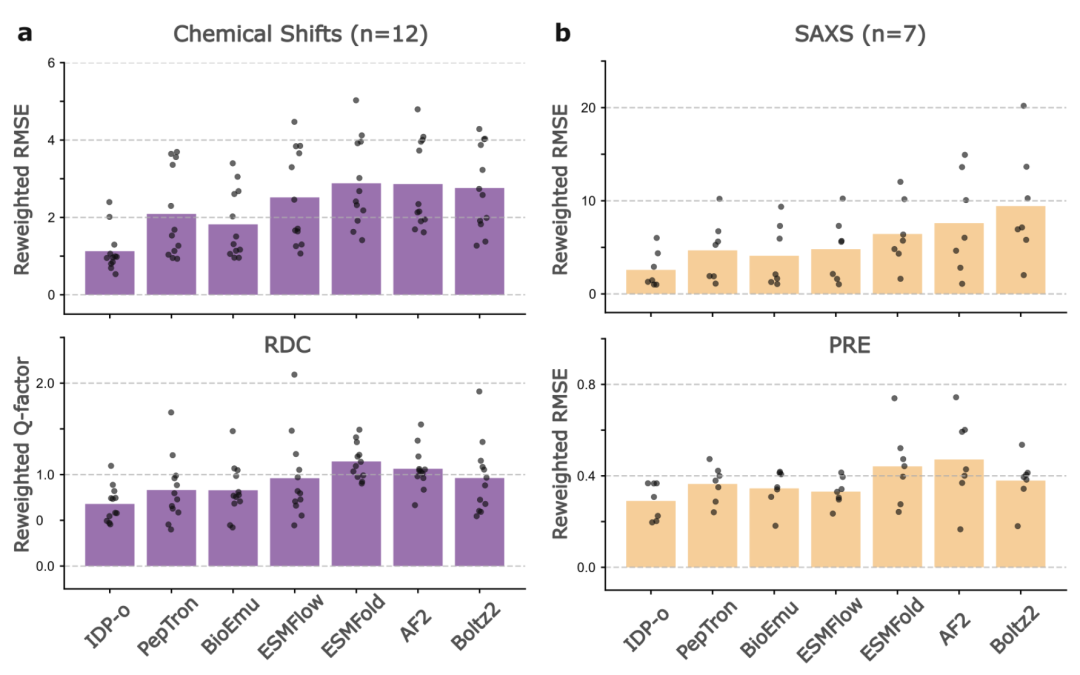
この調査では、現在のモデルに共通するボトルネックもいくつか指摘されています。ほとんどのモデルは長距離接触の好みを捉えることができず、二次構造に様々なバイアスが見られます。さらに、主流のモデルは一般的に「条件付きフォールディング配列」の未フォールディング状態を正確に記述するのに苦労していますが、IDP-oはこの点において独自の優位性を示しています。
無秩序から秩序へ:IDP研究における世界的な進歩と新たな章
本質的に無秩序なタンパク質 (IDP) は、その非常に動的な構造特性と多くの主要な疾患との密接な関連性により、世界中のライフ サイエンスおよび製薬業界における研究の最先端領域として急速に成長しています。
学術界では、AI構造予測技術がIDPの「動的パスワード」を解読する上で重要な力になりつつある。ケンブリッジ大学が提案したAlphaFold-Metainference法は、このアプローチでは、AlphaFold アライメント エラー マップと分子動力学シミュレーションを組み合わせることで、主に安定した構造を予測する従来の AlphaFold の限界を克服し、IDP と無秩序な領域を含む構造セットを正常に構築し、それらの多型性を理解するための新しい方法を提供します。
論文のタイトル:
無秩序タンパク質の構造アンサンブルのAlphaFold予測
論文リンク:https://www.nature.com/articles/s41467-025-56572-9
コペンハーゲン大学のチームは、AlphaFold をタンパク質言語モデルとさらに統合しました。これにより、ヒトの無秩序なプロテオームの立体構造を大規模に予測することが可能になりました。これは、IDP 研究における AI テクノロジーの普遍性と拡張性を実証しています。
論文のタイトル:
ヒトの本質的に無秩序なプロテオームの立体配座集団
論文リンク:https://www.nature.com/articles/s41586-023-07004-5
学術的発見が真に疾患治療を変えることができるかどうかは、産業界が技術を実用化できる能力にかかっています。英国のバイオテクノロジー企業ペプトンとドイツの製薬会社エボテックの提携は…これは、IDP 研究を医薬品開発に拡張するための実現可能な道筋を示しています。Peptone社の超高速水素-重水素交換質量分析(HDX-MS)プラットフォームを活用することで、研究者は無秩序タンパク質の動的変化をリアルタイムで追跡し、従来の構造決定法では同定が困難な結合部位を捕捉することができます。Evotec社の標的検証、薬剤スクリーニング、臨床開発における優位性と組み合わせることで、薬物治療が困難なIDP標的を、薬物候補分子へと転換することが可能になります。
この一連の進歩は、ペプトロンモデルの「秩序構造と無秩序構造の全スペクトルをカバーする」というトレンドを反映するだけでなく、かつては捉えどころのないと考えられていた無秩序タンパク質が、精密医療やバイオ医薬品の重要なターゲットになりつつあることを示しています。継続的な技術革新と産業界の連携の深化により、IDPは将来の疾患治療における理解と介入経路のための全く新しい枠組みを提供する可能性があります。
参考リンク:
1.https://www.vbdata.cn/intelDetail/717834
2.https://c.m.163.com/news/a/JDIR2LQJ0552ZPM2.html
3.https://www.vbdata.cn/intelDetail/580634








