Command Palette
Search for a command to run...
L'université Cornell Propose Un Cadre d'IA Innovant Pour Décoder Le Mécanisme Chimique Des Électrolytes lithium-ion Hautement Conducteurs, Atteignant Un Taux De Réussite De Prédiction Supérieur À 80 % Pour Le %.

Avec l'expansion rapide du marché des batteries à énergie nouvelle, et notamment la généralisation des batteries lithium-ion, des batteries à l'état solide et des batteries à haute densité énergétique, l'optimisation des performances de l'électrolyte est devenue un facteur clé déterminant la sécurité, l'efficacité et la durée de vie des batteries.
La chimie des sels et des solvants sous-tend le comportement des électrolytes dans la plupart des systèmes de batteries lithium-ion, déterminant des propriétés clés telles que la conductivité ionique, la viscosité et la stabilité chimique. Cependant, leur conception rationnelle est contrainte par un vaste espace chimique englobant d'innombrables combinaisons et des relations de couplage non linéaires entre structure et performance. La rareté et l'hétérogénéité des données expérimentales aggravent encore ce problème, limitant la capacité de généralisation des modèles. Ces dernières années, malgré certains progrès dans la découverte autonome d'électrolytes pilotée par l'IA,Cependant, les recherches existantes manquent encore clairement d'un paradigme informatique unifié capable de maintenir une interprétabilité intrinsèque tout en explorant le vaste espace chimique couvert par les formulations d'électrolytes à grande échelle.
Dans ce contexte,Une équipe de recherche de l'Université Cornell a développé SCAN, un cadre robuste, interprétable et efficace en matière de données, pour la modélisation et l'interprétation de la chimie des sels et des solvants.Ce cadre de travail gère efficacement les données à longue traîne et couvre l'ensemble des formulations sel-solvant. Les chercheurs ont appliqué SCAN aux systèmes d'électrolytes non aqueux (NAE), obtenant une erreur de référence de 0,372 mS·cm⁻¹ dans la prédiction de la conductivité, soit une réduction de l'erreur de prédiction de 65,31 TP³T par rapport au modèle de référence.
Plus important encore, une validation à grande échelle montre queLe modèle a atteint un taux de réussite de prédiction de 81,08% pour les systèmes candidats les mieux classés.En plus de ses capacités prédictives, SCAN révèle le mécanisme chimique en introduisant le découplage de gradient, la régression symbolique et les calculs de chimie quantique pour révéler l'influence de la flexibilité moléculaire et des interactions ion-solvant sur la conductivité.
Les résultats de recherche associés, intitulés « Un cadre interprétable guidé par le routage dynamique pour la chimie sel-solvant », ont été publiés dans Nature Computational Science.
Points saillants de la recherche :
* SCAN comble une lacune importante dans la recherche sur la chimie des solvants salins NAE à haute performance.
Inspirés par le modèle de surface d'énergie potentielle centré sur l'atome, nous avons développé un réseau multi-caractéristiques (MFNet) avec une représentation dédiée centrée sur les descripteurs et un mécanisme d'attention.
* De manière novatrice, une stratégie de routage dynamique est introduite dans MFNet, permettant au modèle de prédire avec précision la conductivité ionique sur une large plage sans altérer la distribution des données d'origine.

Adresse du document :
https://www.nature.com/articles/s43588-026-00955-5
Suivez notre compte WeChat officiel et répondez « SCAN » en arrière-plan pour obtenir le PDF complet.
Les dimensions des données sont largement couvertes
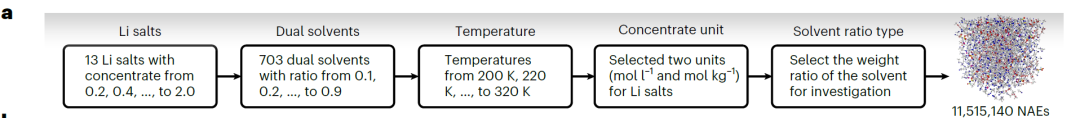
Afin de former un modèle SCAN de haute précision,L'équipe de recherche a construit l'ensemble de données CALiSol, qui comprend 13 sels de lithium et 38 solvants organiques (comme indiqué dans la figure ci-dessous), totalisant 13 302 entrées de données complètes.
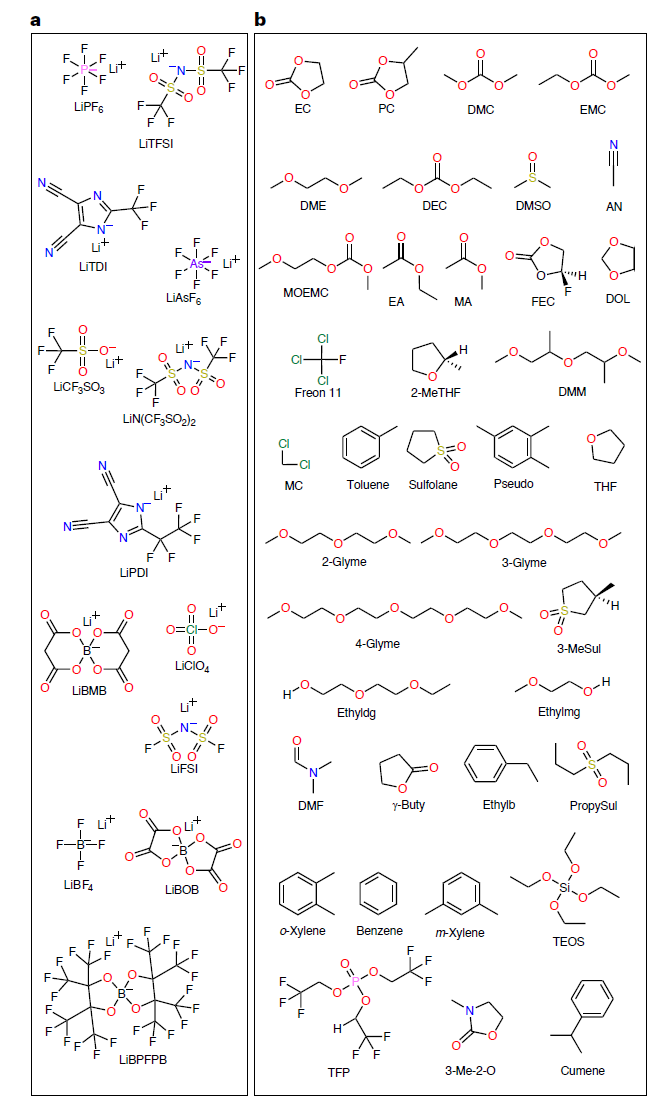
Les dimensions des données sont importantes, et chaque point de données contient les éléments suivants :
Conductivité ionique k : 0–38,1 mS/cm
* Température T : 194,15–477,42 K
* Concentration en sel c : 0–4 mol/L ou mol/kg
* Types de sels de lithium : LiPF₆, LiTFSI, LiFSI, LiBOB, etc., soit un total de 13 types.
* Types de solvants : y compris le carbonate d'éthylène (EC), le carbonate de méthyléthyle (EMC), le propionitrile (AN) et bien d'autres.
* SRT à stratégie mixte : rapport molaire, volumique ou massique
Les informations moléculaires de tous les sels de lithium et solvants ont été converties en coordonnées moléculaires tridimensionnelles à l'aide de séquences SMILES, et une optimisation géométrique a été réalisée au niveau théorique B3LYP/6-31G afin de garantir une structure moléculaire et des propriétés électroniques précises et fiables. Grâce à cette méthode,L'ensemble de données fournit non seulement les valeurs de conductivité ionique, mais aussi les caractéristiques moléculaires, les informations structurelles et l'environnement de solvatation de chaque système.Fournir des données d'entrée riches pour les modèles d'IA, en équilibrant l'intégrité des données et l'interprétabilité scientifique.
Lors de la construction de l'ensemble de données, l'équipe de recherche a accordé une attention particulière au problème des données à longue traîne (LTD) : le nombre de systèmes hautement conducteurs est limité, tandis que la conductivité de la plupart des systèmes est faible - k < 5 mS·cm⁻¹ pour 9 115 NAE (environ 68,5%), et k > 20 mS·cm⁻¹ pour seulement 67 (environ 0,5%).
SCAN : Utilise MFNet et des mécanismes de routage dynamiques
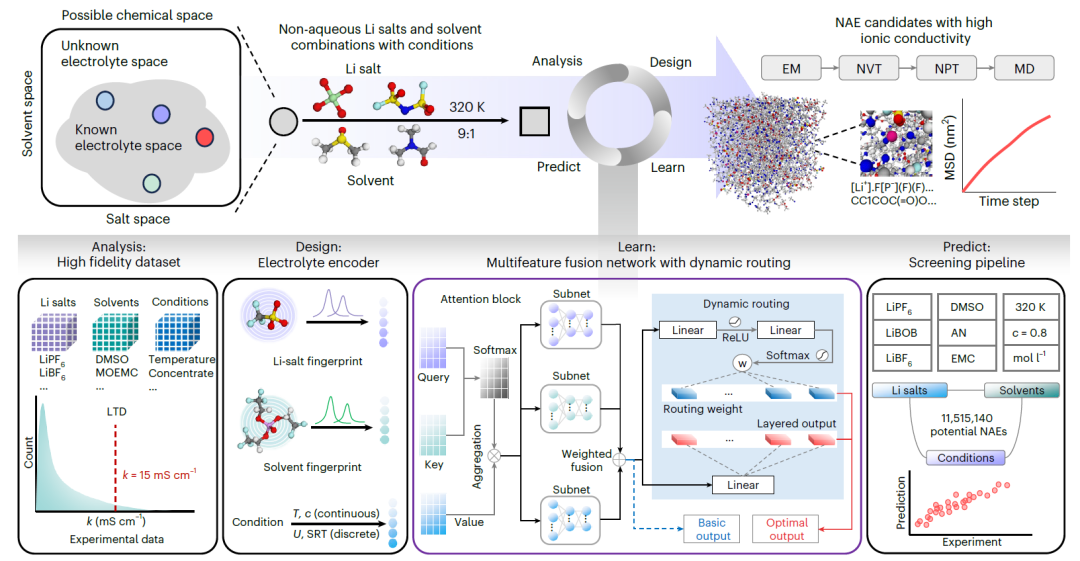
SCAN est une plateforme d'ingénierie NAE guidée par un routage dynamique. Le diagramme suivant illustre son flux de travail complet :
Analyse
Un jeu de données expérimentales de haute fidélité a été constitué, couvrant diverses combinaisons de sels de lithium, de solvants et de conditions expérimentales. Une analyse statistique a été réalisée sur chaque variable, incluant la caractérisation de la distribution à longue traîne des valeurs de k.
Conception
Des empreintes moléculaires contenant des informations chimiques ont été conçues respectivement pour les sels de lithium et les solvants, et combinées à un codage conditionnel pour caractériser avec précision les combinaisons sel-solvant-condition.
Apprendre
Un réseau de fusion multi-caractéristiques personnalisé (MFNet) a été développé pour prédire la valeur k des NAE. Des modules d'attention indépendants ont été construits en intégrant les données de requête, de clé et de valeur afin de représenter respectivement le sel, le solvant et la condition, puis fusionnés à l'aide d'un réseau neuronal entièrement connecté. De plus, une stratégie de routage dynamique a été introduite pour améliorer les performances et la robustesse du modèle.
Prédire
Un système de criblage à haut débit a été mis en place pour identifier les NAE potentiels parmi plus de dix millions de combinaisons sel-solvant. Les systèmes candidats présentant des valeurs k prédites élevées ont ensuite été validés par des simulations de dynamique moléculaire (DM), incluant la minimisation d'énergie (EM), l'ensemble canonique (NVT), l'ensemble isotherme-isobare (NPT), des simulations de production DM, l'analyse du déplacement quadratique moyen (MSD) et le calcul de la fonction de distribution radiale (RDF).
MFNet et le mécanisme de routage dynamique constituent le cœur de l'ensemble du cadre, et les mécanismes spécifiques sont les suivants :
MFNet : Réseau d’auto-attention multicanal
S'inspirant du modèle de surface de potentiel central atomique, le cadre MFNet (Molecular Feature Network) divise le réseau en trois sous-réseaux indépendants, chacun gérant des fonctions différentes :
* Sous-réseau de sel de lithium : Dans l’ensemble de données CALiSol, chaque NAE ne contient qu’un seul type de sel de lithium, son vecteur descripteur est donc directement entré dans le sous-réseau de sel de lithium.
* Sous-réseau de solvants : étant donné que certains points de données impliquent plusieurs solvants (par exemple, PC et AN mélangés dans un rapport de 0,9:0,1), la valeur moyenne de ces descripteurs de solvants est calculée comme entrée pour refléter l’environnement global du solvant.
* Sous-réseau conditionnel : gère les conditions expérimentales telles que la température et la concentration.
Après le module d'auto-attention, deux couches cachées entièrement connectées sont utilisées pour la projection progressive et la transformation non linéaire de l'entrée, générant des caractéristiques de haute dimension afin d'améliorer l'expressivité du traitement ultérieur. La fonction d'activation non linéaire ReLU est utilisée entre les couches pour optimiser la capacité de représentation du modèle.
L'architecture finale du réseau est la suivante : un sous-réseau pour le sel de lithium (14-16-16), un sous-réseau pour le solvant (14-16-16) et un sous-réseau conditionnel (6-16-16). Les sorties potentielles, de dimension intrinsèque (dimensions des caractéristiques < 128), sont traitées par un module de fusion pondérée afin de calculer les sorties pondérées tout en préservant les informations de dépendance globale des caractéristiques d'entrée. Le codage des caractéristiques du sel de lithium, du solvant et des caractéristiques conditionnelles étant traité indépendamment par trois sous-réseaux, l'auto-attention à une seule tête (SHA) peut être utilisée pour traiter légèrement les données de caractéristiques à petite échelle (14, 14 et 6 dimensions, respectivement), ce qui la rend particulièrement adaptée aux tâches nécessitant de faibles dimensions d'intégration.
Mécanisme de routage dynamique : résoudre le problème des données de longue traîne
Dans une distribution à longue traîne, les échantillons de données sont principalement concentrés dans la « tête », tandis que ceux de la « queue » sont rares. Or, les modèles traditionnels ont souvent tendance à surajuster les échantillons de tête et à ignorer ceux de la queue, ce qui limite l'exploration de cet espace chimique pourtant crucial. Pour pallier ce problème, cette étude introduit une stratégie de routage dynamique dans MFNet. Contrairement à l'architecture standard qui traite tous les échantillons de manière égale,Le routage dynamique apprend un mécanisme de contrôle souple pour attribuer de manière adaptative différentes capacités de représentation aux différentes couches en fonction de l'entrée de la couche.Comme indiqué ci-dessous :

Ce mécanisme permet aux échantillons rares d'activer différents chemins de routage et de faciliter le calcul conditionnel, améliorant ainsi la capacité de généralisation pour les catégories peu fréquentes. Ses deux principales caractéristiques sont :
* Poids de routage dépendants de l'entrée : c'est-à-dire la sélection d'un sous-espace de caractéristiques pour chaque échantillon sans modifier la fonction de perte d'origine ni la distribution des données ;
* Découplage des fonctionnalités adaptatif à la catégorie : cela implique de modéliser explicitement la différence entre les catégories dominantes et les catégories de queue, démontrant ainsi que le routage dynamique offre une solution LTD plus flexible et interprétable qu’un simple remodelage statique des pertes.
Interprétabilité : GBA et régression symbolique
SCAN offre une interprétabilité chimique tout en prédisant avec précision la conductivité ionique :
* GBA (Attribution basée sur le gradient) :Le modèle SCAN, reposant sur une architecture de réseau neuronal à trois niveaux d'attention parallèles, présente un processus de décision plus complexe à visualiser que les modèles arborescents. Afin d'identifier les principaux facteurs chimiques influençant k, la méthode GBA a été utilisée pour évaluer l'importance des caractéristiques : elle a permis de calculer la contribution du gradient de chaque caractéristique d'entrée à la sortie du modèle et d'identifier les caractéristiques les plus critiques (sel de lithium, solvant et caractéristiques conditionnelles).
* Régression symbolique :Pour découvrir une relation fonctionnelle interprétable entre les informations clés sur le sel de Li, le solvant et les conditions et kkk, une méthode de régression symbolique basée sur PySRRegressor a été utilisée.
Le modèle SCAN surpasse systématiquement tous les modèles de référence.
Pour évaluer de manière exhaustive les performances du cadre SCAN utilisant MFNet et le routage dynamique, les chercheurs ont construit quatre modèles de base basés sur MFNet.Aucun d'eux ne contient de routage dynamique : (1) Modèle MFNet de base : B1 (descripteur non traité) et B2(Descripteur mis à l'échelle par la valeur maximale) ; (2) Modèle MFNet combinant la technique de suréchantillonnage de la classe minoritaire SMOTE : S1–S18(3) Modèle MFNet combinant la technique de sous-échantillonnage des k plus proches voisins (KUTE) : U1–U9(4) Modèle MFNet combinant SMOTE et KUTE : SU1–SU9 .
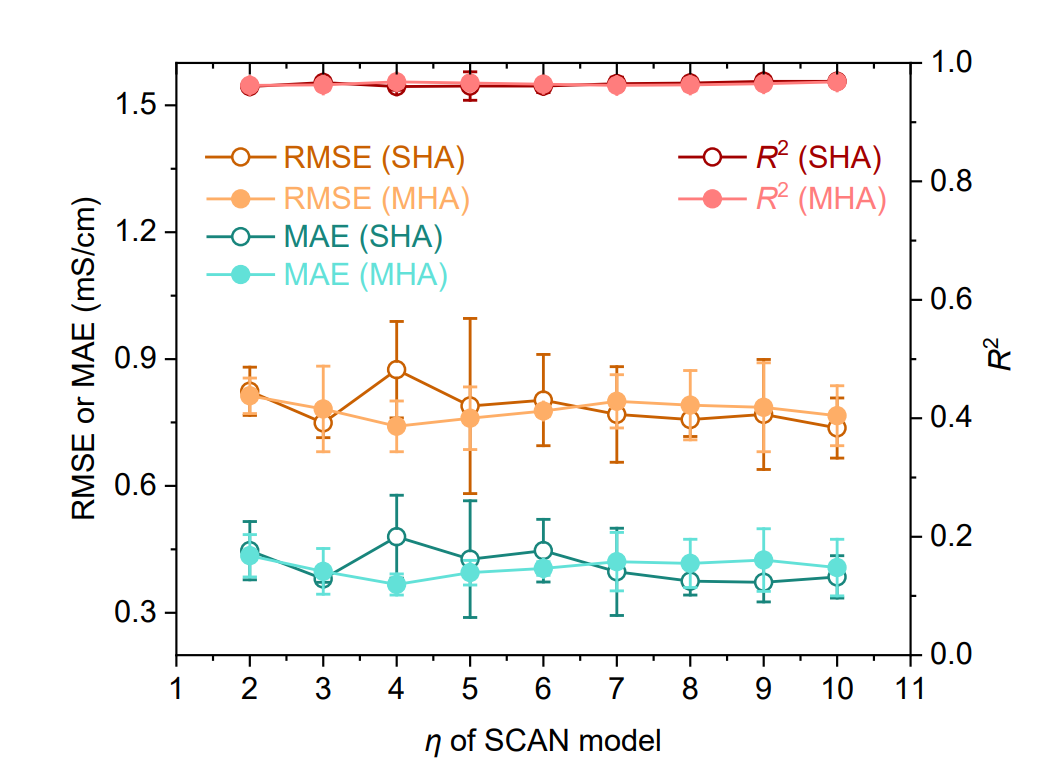
Simultanément, des modèles basés sur l'architecture Transformer, des réseaux de neurones graphiques (GNN), la régression à vecteurs de support (SVR) et l'algorithme XGBoost ont été utilisés comme références, comme illustré dans la figure ci-dessous. De plus, des modèles SCAN utilisant des structures d'attention à une seule tête (SHA) ou à plusieurs têtes (MHA) et différents nombres de couches de routage dynamique (η = 2–10) ont été implémentés afin de vérifier la robustesse du modèle.
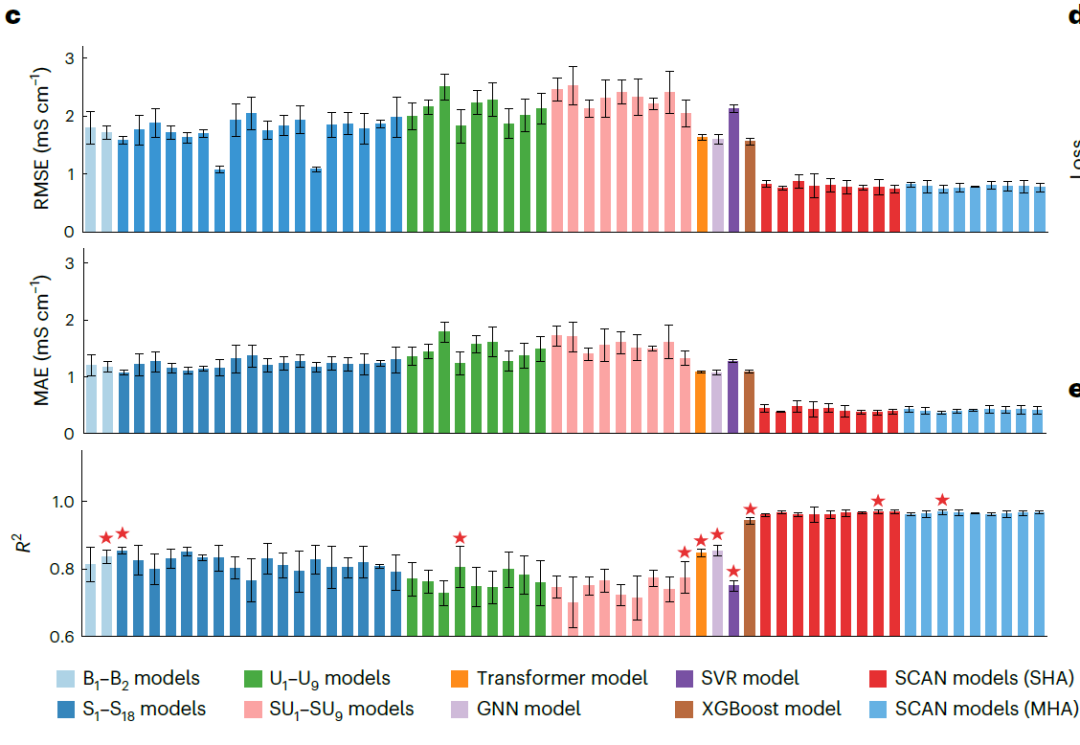
Évaluation des performances de l'évaluation des performances de l'AE
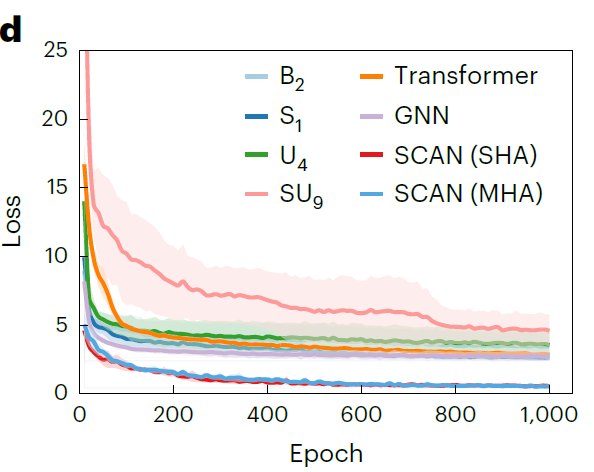
Les résultats présentés dans la figure ci-dessous démontrent que le modèle SCAN utilisant SHA (η = 9) surpasse systématiquement tous les modèles de référence, affichant des erreurs de prédiction significativement plus faibles (RMSE 0,769 mS·cm⁻¹, MAE 0,372 mS·cm⁻¹) et un R² plus élevé (0,969). Il convient de noter que…Comparé au GNN de référence le plus performant (1,072 mS·cm⁻¹) en MAE, l'erreur a été réduite de 65,31 TP3T.Les performances SCAN utilisant MHA (η = 4) sont comparables à celles de la version SHA, ce qui indique que MFNet possède une bonne robustesse dans l'intégration avec le routage dynamique.
aussi,Les chercheurs ont tracé les courbes de perte de validation de SCAN et d'un modèle de référence représentatif sur 1 000 époques d'entraînement.Comme le montre la figure ci-dessous, dès le début de l'entraînement, l'erreur de prédiction de SCAN est significativement inférieure à celle du modèle de référence, avec une perte initiale de seulement 4,59 mS·cm⁻¹, et sa perte de validation continue de diminuer et converge vers un niveau encore plus bas, indiquant qu'il possède une stabilité et une capacité de généralisation plus fortes.
Capacité de criblage NAE à haut débit
Le balayage de haute précision permet d'explorer efficacement le vaste espace chimique des NAE. À cette fin, les chercheurs ont mis au point un processus de criblage à haut débit (illustré dans la figure ci-dessous) conçu pour identifier les NAE à constante diélectrique élevée.Sur cette base, 11 515 140 NAE à double solvant potentiels ont été générés.
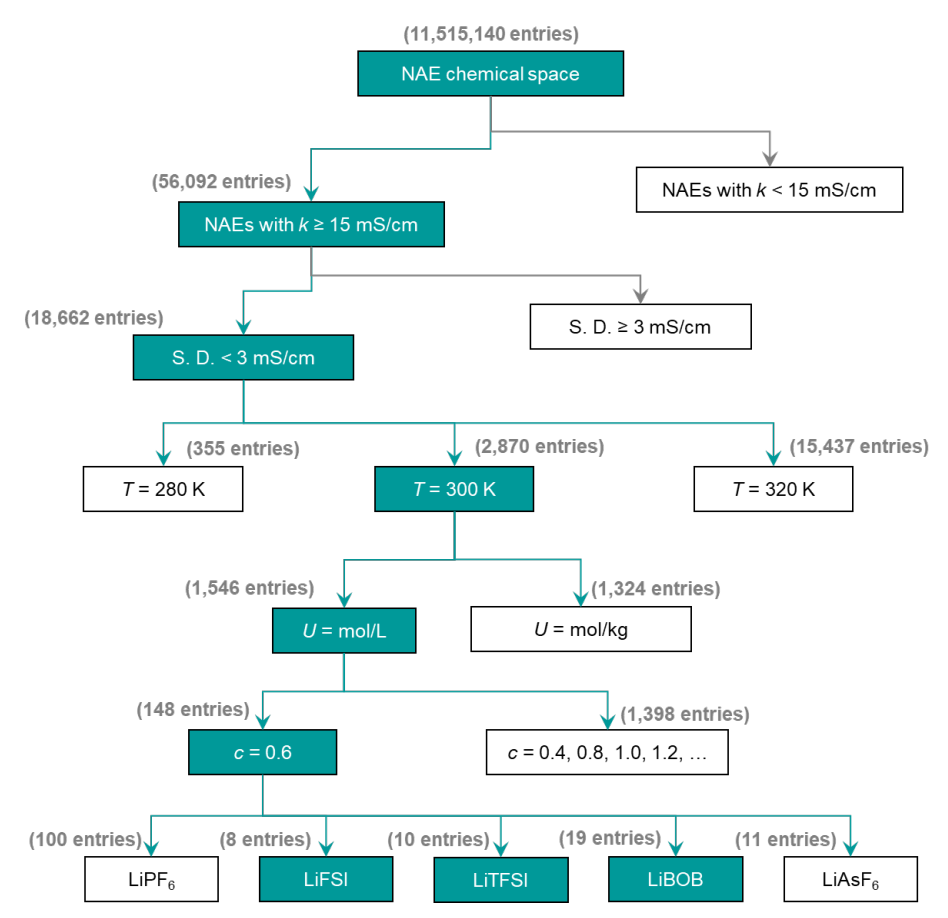
En exploitant la puissance prédictive de SCAN, 56 092 NAE avec k ≥ 15 mS·cm⁻¹ ont été rapidement identifiés, dont 18 662 avec une faible incertitude de prédiction (< 3 mS·cm⁻¹). Ce processus réduit considérablement la charge de calcul et le coût des simulations et expériences de dynamique moléculaire.
Validation des électrolytes non aqueux candidats (NAE)
Afin de valider rigoureusement les prédictions, les chercheurs ont effectué des simulations de dynamique moléculaire (DM) pour obtenir la valeur de k. En fonction de la température (T), de la concentration (c) et des conditions réelles, ils ont restreint le champ de validation DM aux systèmes candidats NAE basés sur LiFSI, LiTFSI et LiBOB, sélectionnant finalement 37 systèmes prometteurs pour une étude détaillée, comme illustré dans la figure ci-dessous :
Le temps de calcul moyen pour chaque système est d'environ 10 heures (36 355,12 secondes), et le coût estimé de la simulation de 10⁷ systèmes candidats est d'environ 10⁸ heures GPU, ce qui dépasse largement les capacités pratiques d'une sélection méthodologique par force brute, même dans des conditions idéales de calcul haute performance. En comparaison,Un modèle SCAN bien entraîné peut effectuer l'ensemble du processus, du calcul du descripteur à la prédiction finale, en moins de 5 secondes pour chaque système candidat, réduisant ainsi les coûts de calcul de plus de 7 200 fois.Cela améliore considérablement l'évolutivité et l'efficacité. Ceci souligne la nécessité du cadre SCAN dans la découverte de NAE, dont le modèle de substitution permet de prioriser rapidement les candidats les plus performants à un coût de calcul extrêmement faible.
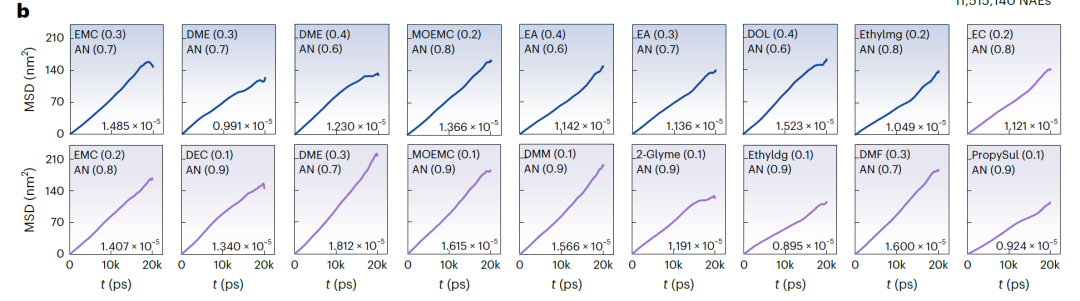
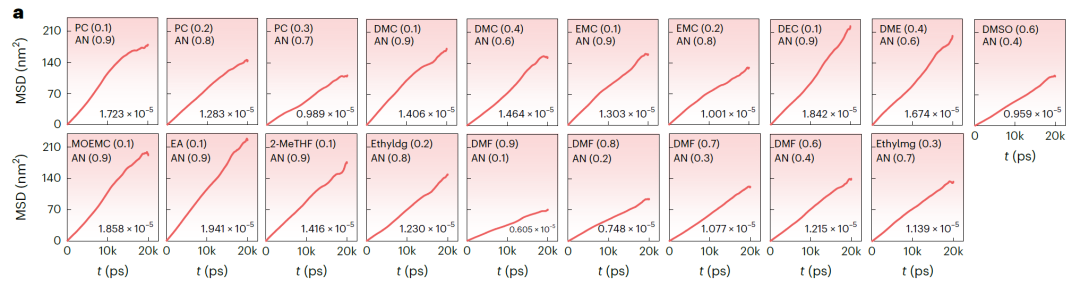
Les figures b (NAE basées sur LiFSI et LiTFSI) et a (NAE basées sur LiBOB) ci-dessous présentent le déplacement quadratique moyen (DQM) au cours du temps et le coefficient de diffusion correspondant, obtenus par simulations de dynamique moléculaire (DM). Le DQM de tous les systèmes présente une augmentation linéaire stable, indiquant la stabilité et la convergence de la simulation. Les valeurs de k prédites par SCAN sont très cohérentes avec les résultats de la simulation DM, avec un écart moyen de seulement 3,198 mS·cm⁻¹ et un écart maximal de 7,342 mS·cm⁻¹. Dans les systèmes de validation : pour 25 systèmes avec k > 15 mS·cm⁻¹, le taux de réussite était de 67,571 TP⁻¹. Pour les systèmes avec k prédit > 14 mS·cm⁻¹, le taux de réussite de la validation a atteint 81,081 TP⁻¹.
L'intelligence artificielle est en train de remodeler le paradigme fondamental de la recherche et du développement des batteries.
Les batteries, composants essentiels d'applications clés telles que l'électronique grand public, les véhicules électriques et les systèmes de stockage d'énergie pour le réseau électrique, sont un moteur de la transition énergétique mondiale. Améliorer la densité énergétique et la puissance, prolonger la durée de vie, renforcer la sécurité et réduire les coûts de fabrication sont les principaux objectifs de la recherche et du développement des batteries. La clé de la réalisation de ces objectifs réside dans une compréhension approfondie des mécanismes électrochimiques au sein des batteries, notamment les réactions d'interface électrochimiques et les mécanismes de stabilisation, le couplage et le transport des électrons et des ions, ainsi que les mécanismes de stockage d'énergie des matériaux d'électrode de nouvelle génération.
Dans une perspective plus large d'évolution technologique, le paradigme « modélisation interprétable et axée sur les données », représenté par le cadre SCAN, devient un élément essentiel du système de R&D des batteries de nouvelle génération. Ces dernières décennies, l'innovation dans les matériaux pour batteries s'est principalement appuyée sur l'expérience et des expérimentations empiriques, engendrant des cycles de développement longs et des coûts élevés. Cependant, grâce à l'intégration de l'apprentissage automatique et du calcul à haut débit, la R&D des batteries gagne en efficacité.
Par exemple,Wen Yan et Sheng Gong, entre autres membres de l'équipe Seed de ByteDance, ont développé un cadre unifié pour la conception de formulations d'électrolytes qui intègre des modèles de prédiction directe avec des méthodes de génération inverse.Des chercheurs ont collecté un grand nombre de données bibliographiques sur des molécules individuelles (plus de 240 000) et des mélanges moléculaires (plus de 10 000) avec des étiquettes de propriétés, couvrant ainsi un large éventail de possibilités en matière de conception d'électrolytes. En intégrant ensuite plus de 100 000 données de mélanges moléculaires issues de simulations de dynamique moléculaire, ils ont pu entraîner un modèle d'apprentissage automatique précis, capable de prédire non seulement la conductivité, mais aussi les structures de fusion liées à la stabilité interfaciale des batteries au lithium métal.
Titre de l'article : Une solution unifiée, prédictive et générative, pour la formulation d'électrolytes liquides
Lien vers l'article :
https://www.nature.com/articles/s42256-025-01173-w
De manière générale, l'intelligence artificielle redéfinit le paradigme fondamental de la recherche et du développement des batteries. Grâce à une intégration poussée des modèles et des systèmes expérimentaux, la vitesse de découverte et la densité d'innovation des matériaux pour batteries devraient connaître une nouvelle accélération.
Références :
https://www.nature.com/articles/s43588-026-00955-5
https://phys.org/news/2026-02-ai-framework-reveals-chemistry-high.html
https://static-content.springer.com/esm/art%3A10.1038%2Fs43588-026-00955-5/MediaObjects/43588_2026_955_MOESM1_ESM.pdf
https://www.eet-china.com/mp/a471613.html








