Command Palette
Search for a command to run...
En Seulement 30 Minutes, l'agent Biologique multi-agent Robin a Intégré Avec Succès 550 Articles De Recherche, Établissant Une Boucle De Recherche Autonome Et Identifiant Des Thérapies Candidates Pour La DMLA diabétique.

Grâce à la maturation continue des technologies de détection biologique, d'expérimentation sur les perturbations et de modélisation informatique, la précision et l'échelle de la recherche en sciences de la vie progressent rapidement. Cependant, comparées à l'augmentation exponentielle des capacités de production de données, les capacités d'intelligence des systèmes de recherche en matière d'intégration des connaissances et de raisonnement scientifique restent nettement en deçà.Une quantité considérable d'informations précieuses est dispersée dans des articles, des bases de données et des résultats expérimentaux. Le tri manuel est non seulement inefficace, mais il complique également la mise en relation des conclusions existantes entre différents domaines.Il en résulte que de nombreuses découvertes validées n'ont pas pu être traduites en temps opportun en nouvelles idées de recherche ou en protocoles cliniques.
Ce problème de « fragmentation des connaissances » est particulièrement évident dans le domaine du « repositionnement des médicaments ». Qu'il s'agisse de la découverte tardive des effets otoprotecteurs du dabrafénib ou de l'élargissement du champ d'application thérapeutique de la kétamine, les deux ont connu des retards de transfert de plusieurs années, voire de plusieurs décennies, reflétant les goulots d'étranglement actuels dans la découverte et l'intégration des connaissances au sein du processus de recherche scientifique.
Ces dernières années, les grands modèles de langage (LLM), grâce à leurs capacités de recherche, d'induction et de raisonnement logique développées par l'entraînement sur des corpus massifs, ont commencé à démontrer leur potentiel dans la recherche en sciences de la vie. En combinant le réglage fin, la génération améliorée par la recherche (RAG) et les techniques de collaboration multi-agents, ces modèles ont atteint, voire dépassé, les performances humaines dans des tâches individuelles telles que l'analyse de la littérature, la prédiction de médicaments et la formulation d'hypothèses scientifiques. Cependant,La plupart des outils d'IA existants ne couvrent encore que certaines parties du processus de recherche et ne peuvent pas véritablement relier l'ensemble de la chaîne « génération d'hypothèses – conception expérimentale – analyse des données – itération des résultats ».Par conséquent, une recherche scientifique intelligente en boucle fermée ne peut pas encore être réalisée.
Pour remédier à ce problème, une équipe conjointe de FutureHouse à San Francisco, de l'Université d'Oxford et de l'Université Fordham a proposé le système multi-agents biologiques Robin.Il s'agit du premier système biomédical intelligent qui intègre simultanément les capacités de génération d'hypothèses scientifiques et d'analyse de données expérimentales, et qui réalise un flux de travail en boucle fermée continue.
Grâce à la collaboration entre un agent de recherche bibliographique et un agent d'analyse de données, Robin peut réaliser de manière semi-autonome l'analyse des mécanismes pathologiques, le criblage de candidats médicaments, l'examen expérimental et l'itération d'hypothèses. L'équipe de recherche a utilisé la dégénérescence maculaire liée à l'âge (DMLA) sèche, une maladie aux options thérapeutiques limitées et aux besoins cliniques urgents, comme cas d'application pour valider les capacités de Robin en matière de criblage intelligent de médicaments. Cette approche offre un nouveau paradigme pratique pour le développement de nouveaux médicaments et le repositionnement de médicaments pilotés par l'IA.
Les résultats de cette recherche, intitulée « Un système multi-agents pour l'automatisation de la découverte scientifique », ont été publiés dans la revue Nature.
Points saillants de la recherche :
* Le système Robin est le premier à intégrer la génération d'hypothèses issues de la littérature et l'analyse de données expérimentales biologiques dans un flux de travail continu en boucle fermée.
Robin s'adapte aux résultats de la recherche scientifique multidisciplinaire. Dans le domaine du développement de médicaments thérapeutiques, il suffit de saisir le nom de la maladie ciblée pour que le système puisse automatiquement identifier les mécanismes pathologiques clés, les associer à des modèles expérimentaux in vitro, proposer des médicaments candidats, analyser les données expérimentales et mettre à jour de manière itérative les molécules candidates.
En utilisant la DMLA sèche comme exemple de recherche, Robin a d'abord proposé une nouvelle stratégie pour traiter la dégénérescence maculaire sèche en utilisant des inhibiteurs de ROCK pour améliorer la fonction phagocytaire de l'EPR.

Voir le document :
https://www.nature.com/articles/s41586-026-10652-y
Jeux de données : couvrant la littérature publique, les benchmarks bioinformatiques et les données expérimentales
Cette étude a construit un système de données à trois niveaux comprenant des données issues de la littérature publique, des données de référence générales en bioinformatique et des données auto-expérimentales.Il couvre différents types de tâches, notamment les textes littéraires, les tâches d'analyse bioinformatique, la détection cellulaire et le séquençage du transcriptome.Il couvre essentiellement les principaux scénarios de données dans le processus de développement de médicaments basés sur l'IA.
Dans un premier temps, les chercheurs ont intégré 551 articles de recherche scientifique chinois et anglais relatifs à la DAMD comme base de connaissances pour le système afin de générer des hypothèses scientifiques.Cela comprend 151 études sur les mécanismes des maladies et 400 articles de recherche sur la fonction phagocytaire des cellules de l'épithélium pigmentaire rétinien et son association avec les maladies.Cette littérature permet non seulement d'élucider les mécanismes pathologiques, mais aussi de fournir un cadre théorique pour le criblage de modèles expérimentaux in vitro et l'identification de candidats médicaments en vue de leur repositionnement. Elle constitue la principale source des travaux de recherche de Robin.
Deuxièmement, les chercheurs ont utilisé l'ensemble de données de référence en bioinformatique BixBench pour évaluer quantitativement les capacités d'analyse de données du système.L'étude a sélectionné 170 questions d'examen liées au développement de médicaments.Il couvre différents types de tâches, notamment l'analyse du transcriptome, la génomique, l'analyse d'enrichissement fonctionnel, l'analyse de séquences et les tests statistiques. Toutes les questions sont accompagnées de jeux de données standardisés, de réponses types et de distracteurs, permettant d'évaluer systématiquement l'adaptabilité et la stabilité des agents dans des scénarios bioinformatiques réels.
aussi,Les chercheurs ont également constitué leur propre ensemble de données expérimentales afin de fournir un support concret pour l'itération du modèle et la vérification expérimentale.Les données comprennent les résultats de cytométrie en flux des cellules ARPE-19 et des cellules souches épithéliales pigmentaires rétiniennes humaines primaires, les données de transcriptome par RNA-seq après différents traitements médicamenteux, ainsi que les résultats de cytotoxicité, de coloration immunocytochimique et de dosage immuno-enzymatique (ELISA) du VEGF. Les échantillons de cellules humaines proviennent de la banque d'yeux New York Vision Repair et sont tous des cellules souches épithéliales pigmentaires rétiniennes issues de donneurs âgés de 60 ans et plus, exempts de toute pathologie oculaire, garantissant ainsi l'authenticité et la pertinence clinique des données expérimentales.
Robin : Systèmes multi-agents pour la découverte en sciences biomédicales
Robin, basé sur le framework Aviary et fonctionnant dans l'environnement Jupyter Notebook, se distingue des outils d'IA de recherche traditionnels qui n'effectuent que des tâches isolées. Il est le premier à mettre en œuvre un flux de travail continu en boucle fermée : « génération d'hypothèses scientifiques – analyse expérimentale – retour d'information sur les résultats – itération des hypothèses ».Il peut mener à bien, de manière semi-autonome, l'ensemble du processus de recherche scientifique, y compris la recherche sur les mécanismes des maladies, la sélection des médicaments candidats et l'analyse des données expérimentales.
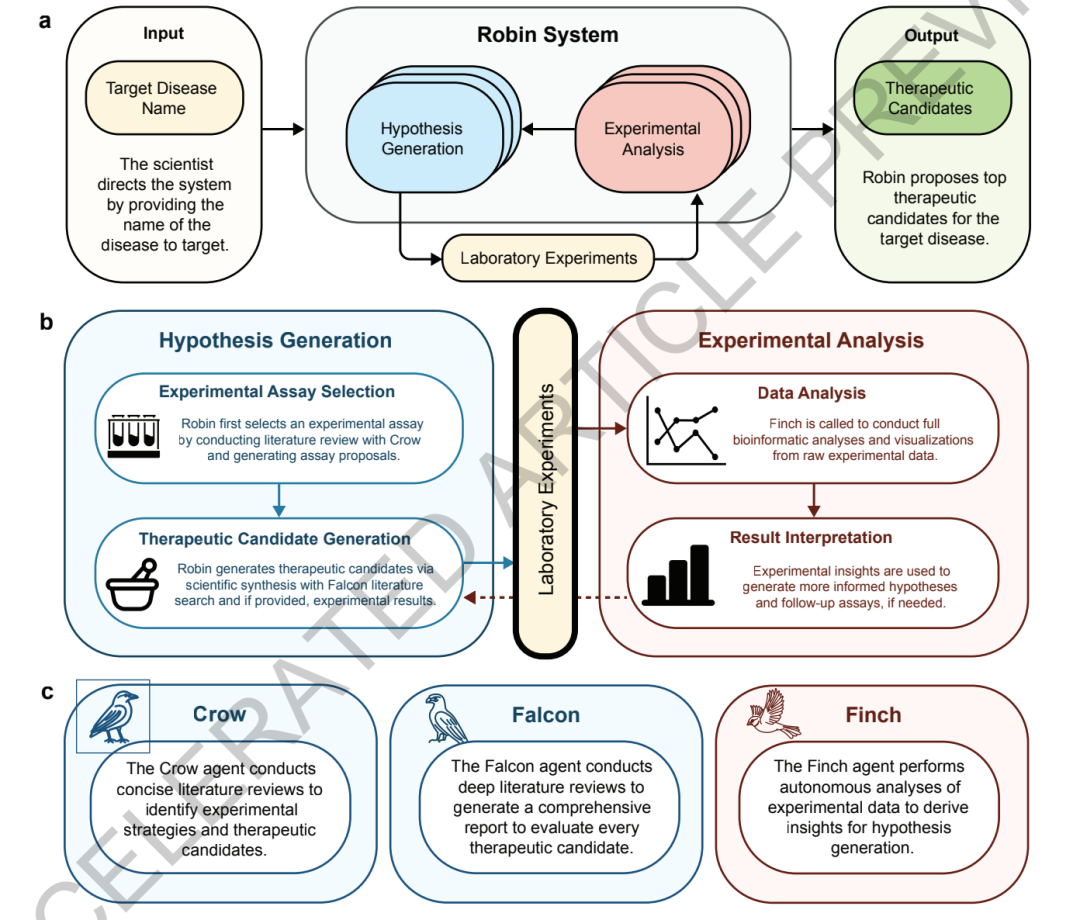
Le système adopte une architecture centrale à « trois agents », composée de deux agents de traitement de documents et d'un agent d'analyse de données travaillant ensemble.
dans,Les deux agents de renseignement documentaire, Crow et Falcon, sont principalement responsables de l'extraction de connaissances documentaires et de la génération d'hypothèses scientifiques.Les deux outils reposent sur le modèle OpenAI o4-mini. Crow se charge de la recherche bibliographique sur les maladies, de l'analyse des mécanismes pathologiques, du criblage des modèles expérimentaux et de la découverte préliminaire de candidats médicaments. Il intègre systématiquement des recherches fragmentées et en extrait les principales conclusions scientifiques. Falcon assure la validation et l'optimisation approfondies. Il analyse plus en détail les mécanismes pharmacologiques, les fondements théoriques et les limitations potentielles des solutions candidates proposées par Crow, et corrige les citations erronées dans la littérature, réduisant ainsi le problème d'« illusion » lié aux grands modèles.
Le troisième module principal, Finch, est un agent intelligent conçu spécifiquement pour l'analyse des données expérimentales biologiques.Contrairement aux outils traditionnels qui utilisent des scripts d'analyse fixes, Finch emploie l'inférence générative, en générant et en exécutant du code Python ou R en temps réel à partir des caractéristiques des données expérimentales. Ceci lui permet d'effectuer de manière adaptative des tâches telles que l'analyse par cytométrie en flux, l'analyse d'expression différentielle RNA-seq et l'enrichissement fonctionnel des gènes. Ainsi, le système n'est plus limité à des flux de travail d'analyse prédéfinis, mais peut ajuster dynamiquement ses stratégies d'analyse, à l'instar d'un chercheur.
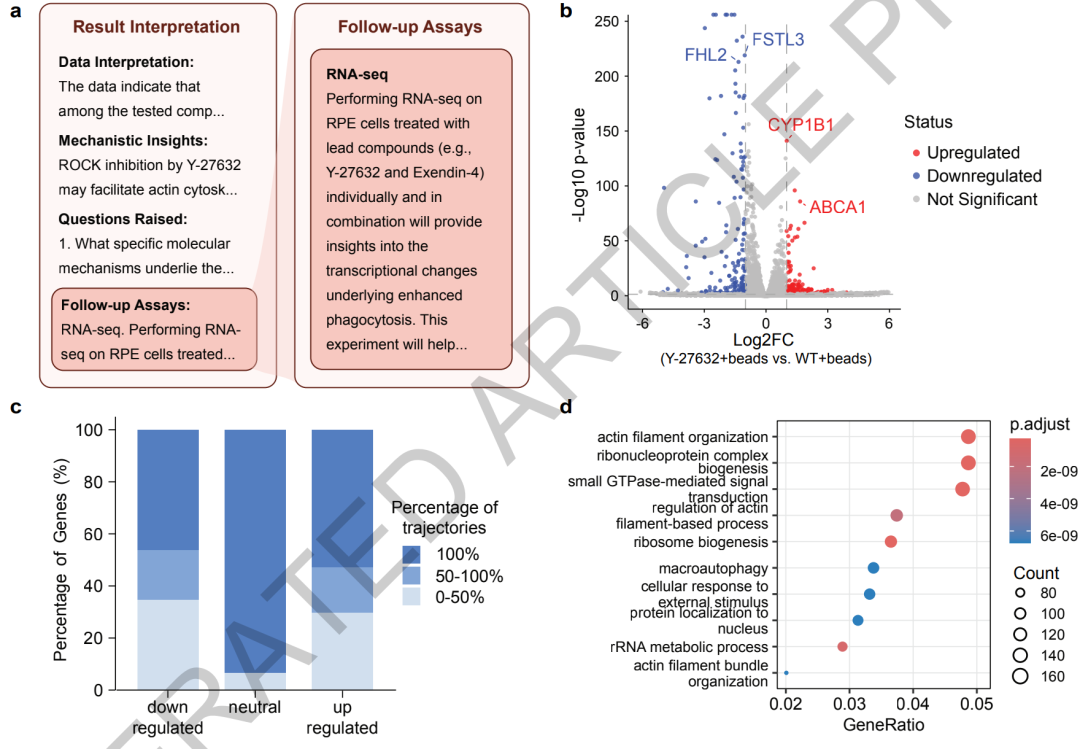
Pour réduire le caractère aléatoire des grands modèles dans l'analyse des données,Robin a par ailleurs conçu un mécanisme d'« analyse multi-trajectoires + intégration consensuelle ».Le système peut lancer simultanément huit trajectoires d'analyse de Finch indépendantes. Chaque trajectoire effectue indépendamment la génération de code, l'analyse des données et la production des résultats. Enfin, les conclusions des différentes trajectoires sont intégrées par méta-analyse, ce qui réduit les écarts dus aux fluctuations et aux différences de paramètres lors d'une analyse individuelle et améliore la stabilité des résultats.
En matière de mécanismes d'évaluation, Robin a également introduit un système d'examen de modèles à grande échelle à deux niveaux.Le système utilise Claude 3.7 Sonnet d'Anthropic comme modèle d'évaluation principal et le combine avec Google Gemini 2.5 Pro pour s'aligner sur les préférences des experts du domaine.Les mécanismes candidats, les modèles expérimentaux et les schémas thérapeutiques sont évalués hiérarchiquement par comparaisons deux à deux et classement par tournoi. Lorsque le nombre de schémas à évaluer est faible, un appariement complet est utilisé ; lorsqu’il est élevé, un échantillonnage aléatoire est employé. Le modèle de Bradley-Terry-Luce est utilisé pour établir le classement pondéré, garantissant ainsi la précision de l’évaluation tout en maîtrisant les coûts de calcul.
De plus, afin de garantir la reproductibilité du processus d'analyse, toutes les tâches Finch s'exécutent dans des environnements conteneurisés Docker indépendants et sont préinstallées avec une chaîne d'outils bioinformatiques complète. L'équipe de recherche a également optimisé et simplifié le flux de travail grâce à plusieurs itérations d'ingénierie Prompt, transformant le processus initial complexe en un flux de travail Jupyter stable et convivial, ce qui améliore encore l'opérabilité du système dans les contextes de recherche.
Robin a découvert que le lipasudil augmente la capacité phagocytaire de 1,89 fois.
Cette étude se concentre sur la DMLA diabétique comme scénario d'application principal et conçoit plusieurs séries d'expériences de validation autour des capacités de génération d'hypothèses, des capacités d'analyse de données, de l'efficacité architecturale et de l'efficacité réelle du développement de médicaments de Robin.
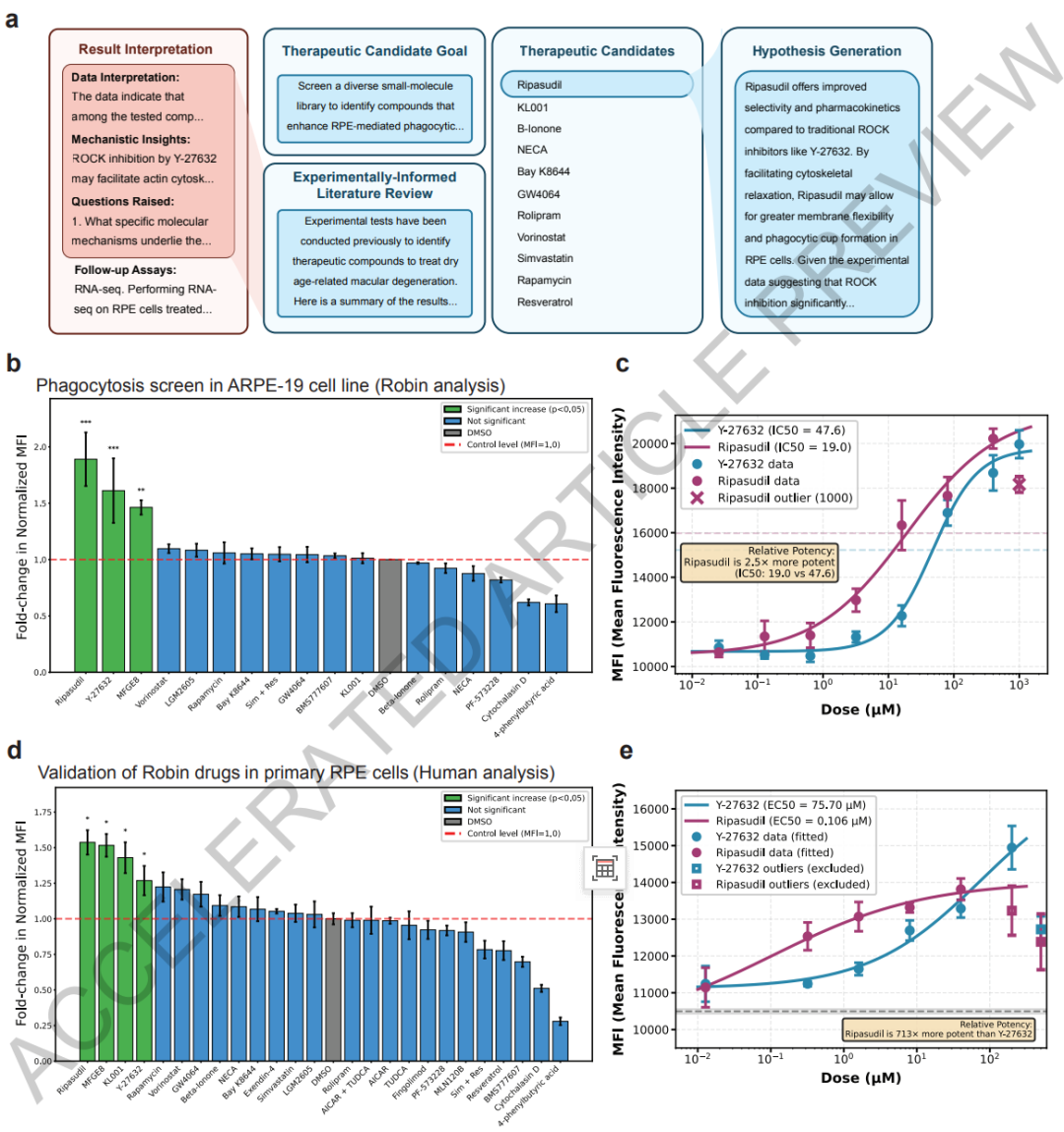
Les expériences principales portent sur le criblage de médicaments candidats et la vérification de leur mécanisme d'action. Robin a d'abord identifié 10 mécanismes pathogènes clés de la DMLA diabétique grâce à une analyse de la littérature et a déterminé que « l'amélioration de la fonction phagocytaire des cellules épithéliales pigmentaires rétiniennes » constituait la principale orientation du traitement.Lors de la première phase de sélection, le système a proposé 30 médicaments candidats. Les chercheurs ont retenu l'exénatide, le fingolimod, le Y-27632 et d'autres médicaments parmi ceux-ci pour mener des expériences, et ont utilisé le médicament efficace connu MFGE8 comme témoin positif.
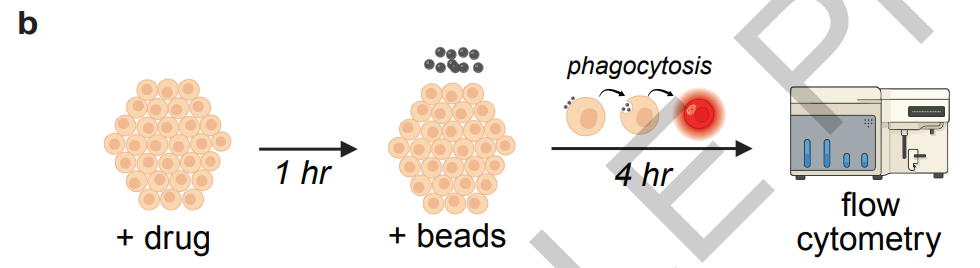
Par la suite, Robin a proposé indépendamment un protocole expérimental de séquençage du transcriptome, et Finch a finalisé l'analyse des données. Les résultats ont montré que le Y-27632 peut reprogrammer le transcriptome des cellules de l'épithélium pigmentaire rétinien en régulant le cytosquelette d'actine, la voie d'autophagie et le gène clé du transport des lipides ABCA1, révélant ainsi un mécanisme d'action jusqu'alors inconnu.
Afin d'améliorer encore la pertinence clinique du criblage de médicaments, l'étude a ensuite mené une deuxième série d'expériences d'itération de médicaments. Robin a ajouté 10 nouveaux candidats médicaments et a constaté que le médicament contre le glaucome commercialisé, le ribasudil, était plus efficace que le Y-27632, augmentant la capacité de phagocytose cellulaire d'environ 1,89 fois.L'équipe de recherche a ensuite utilisé des cellules souches épithéliales pigmentaires rétiniennes primaires humaines, plus proches de l'environnement physiologique réel, pour un nouveau criblage. Les résultats ont confirmé les effets dose-dépendants du ribasudil et du Y-27632 et ont montré que le ribasudil ne présente pas de cytotoxicité notable et possède un fort potentiel pour une application clinique.
Robin a notamment découvert que le régulateur du rythme circadien KL001 possède également le potentiel d'améliorer la fonction phagocytaire, ouvrant ainsi une nouvelle voie de recherche pour le traitement de la DMLA. La validation transcriptomique ultérieure a confirmé que le ripasudil peut réguler positivement et de manière stable l'expression d'ABCA1, clarifiant ainsi son mécanisme d'action principal.
Dans le cadre d'une comparaison avec des systèmes de recherche en IA générale concurrents, l'équipe de recherche a utilisé les mêmes instructions pour appeler l'agent de recherche profonde d'OpenAI. Aucun des 17 médicaments candidats générés n'a montré d'activité d'amélioration de la phagocytose, et aucun n'a permis d'identifier le mécanisme central de l'inhibition de ROCK, ce qui souligne encore davantage les avantages d'adaptabilité de Robin dans des contextes biomédicaux spécifiques.
De plus, dans le test de référence BixBench,La précision globale de l'agent Finch a atteint 22,8±1,7%, ce qui est significativement plus élevé que les 1,6±1,2% du modèle de langage large pur.Les taux de précision pour les tâches de biostatistique ont atteint 47,9 ± 1,51 TP3T, pour l'analyse de cytométrie en flux de base, 1001 TP3T, et pour l'analyse RNA-seq, 861 TP3T. Ces résultats indiquent que le cadre d'agents de recherche spécialement conçu peut améliorer significativement les capacités pratiques des grands modèles à usage général pour l'analyse de données biologiques ; toutefois, des optimisations supplémentaires sont possibles pour les tâches bioinformatiques complexes et multi-étapes.
Robin présente également un avantage certain en termes d'efficacité et de coût. Les statistiques de recherche montrent que le coût moyen d'un flux de travail de recherche complet utilisant ce système n'est que d'environ 10,76 $ ; par ailleurs,Robin a pu réaliser l'analyse intégrée de 551 documents en 30 minutes.Le même volume de travail nécessiterait généralement plus de 800 heures de traitement manuel. Au total, le système effectue un cycle complet de recherche en moins de 2 heures, soit environ 200 fois plus efficacement que les méthodes manuelles traditionnelles.
Derniers mots
L'importance de Robin dépasse la simple découverte de plusieurs candidats médicaments potentiels. Plus important encore, elle démontre pour la première fois le potentiel de l'intelligence artificielle à évoluer d'un « outil d'assistance » à un « système de recherche semi-autonome » en sciences de la vie. Bien sûr, de tels systèmes sont encore loin d'être de véritables « scientifiques autonomes ». Des aspects tels que la conception expérimentale complexe, la compréhension des mécanismes biologiques à différentes échelles et l'interprétabilité des résultats dépendent encore fortement de l'intervention d'experts du domaine. Cependant, l'émergence de Robin montre au moins que l'IA n'est plus seulement un outil destiné à aider les chercheurs à « améliorer leur efficacité », mais qu'elle acquiert progressivement la capacité de participer elle-même à la découverte scientifique.








