Command Palette
Search for a command to run...
À Partir De Données Spectrales Simulées Provenant De 2 000 Matériaux Semi-conducteurs, l'équipe Du MIT a Proposé DefectNet, Capable d'analyser Six Défauts De Substitution coexistants.
En biologie, les défauts sont généralement considérés comme nuisibles. Cependant, en science des matériaux, il est possible de les concevoir intentionnellement afin de conférer aux matériaux des propriétés nouvelles et utiles. Aujourd'hui, des défauts à l'échelle atomique sont introduits avec précision dans les procédés de fabrication de produits tels que l'acier, les semi-conducteurs et les cellules solaires, dans le but d'améliorer leur résistance, de contrôler leur conductivité, d'optimiser leurs performances, et bien plus encore. Par exemple, la concentration de porteurs peut être ajustée par dopage contrôlé dans le silicium ; de même, la manipulation des défauts dans les semi-conducteurs à très large bande interdite peut accroître leur potentiel d'utilisation dans l'électronique de puissance de nouvelle génération.
Bien que l'analyse des défauts soit devenue un outil puissant, mesurer avec précision les différents types et concentrations de défauts dans les produits finis demeure extrêmement complexe, notamment sans découper ni endommager le matériau final. Les ingénieurs qui ignorent les défauts présents dans le matériau risquent de produire des produits aux performances médiocres ou aux caractéristiques non voulues. Malgré l'abondance des techniques de caractérisation des défauts existantes, des limitations importantes persistent en termes de sensibilité, de sélectivité, de quantification et de destructivité.
Dans ce contexte,Une équipe de recherche du MIT a proposé un modèle d'apprentissage automatique fondamental appelé DefectNet, qui peut prédire directement les types chimiques et les concentrations de défauts ponctuels de substitution à partir de spectres vibrationnels mesurés dans la densité d'états de phonons (PDoS).Ce modèle reste opérationnel même en présence de plusieurs éléments. Entraîné sur plus de 16 000 points de données spectrales simulées provenant de 2 000 matériaux semi-conducteurs, il utilise un mécanisme d'attention personnalisé pour identifier jusqu'à six éléments de défauts différents, avec des concentrations allant de 0,21 TP³T à 251 TP³T. Le modèle présente une bonne capacité de généralisation sur des cristaux inconnus contenant 56 éléments et peut être affiné à l'aide de données expérimentales. Sa validation par des données expérimentales de diffusion inélastique d'alliages SiGe et de supraconducteurs MgB₂ démontre sa précision et sa transférabilité.
Les résultats de recherche associés, intitulés « Un modèle de base pour l'identification non destructive des défauts à partir de spectres vibrationnels », ont été publiés en tant que prépublication sur arXiv.
Points saillants de la recherche :
La combinaison de mesures de spectroscopie vibrationnelle de la densité d'états de phonons (PDoS) avec l'apprentissage automatique offre une voie prometteuse pour la caractérisation et la quantification non destructives des défauts ponctuels dans les matériaux massifs.
* Introduction d'un mécanisme d'attention spectrale pour résoudre le problème selon lequel les spectres de phonons des cristaux défectueux et parfaits peuvent être presque indiscernables dans des conditions de faible concentration.
* Introduction des potentiels interatomiques d'apprentissage automatique (MLIP) pour relever le défi des coûts de calcul élevés dans les simulations de phonons basées sur la théorie fonctionnelle de la densité (DFT).

Adresse du document :
https://arxiv.org/abs/2506.00725
Consultez d'autres articles sur l'IA :
Ensemble de données : Un ensemble de données construit à partir de 2 000 matériaux cristallins parfaits
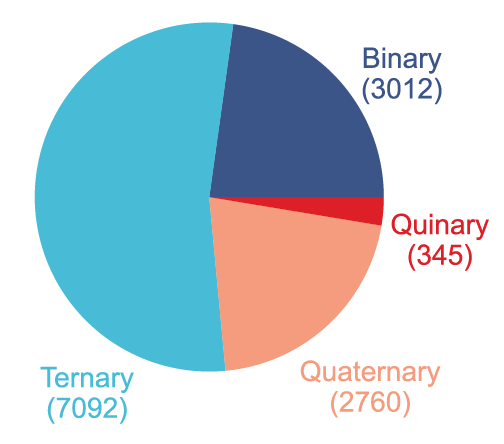
L'étude a constitué un ensemble de données contenant 16 000 supercellules dopées à partir de 2 000 matériaux cristallins parfaits.Il couvre les semi-conducteurs binaires, ternaires, quaternaires et pentaniques, comme illustré dans la figure ci-dessous :
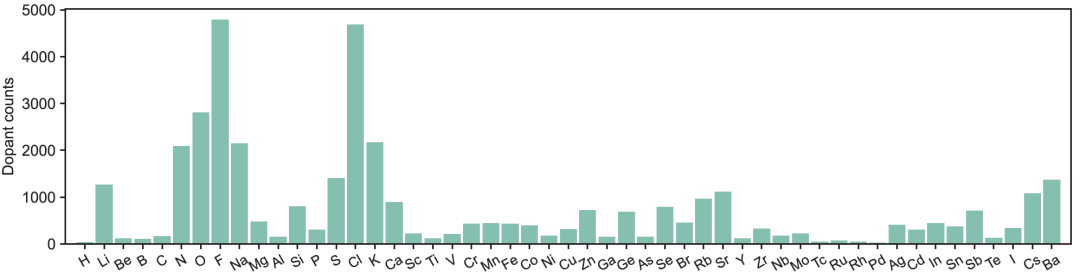
Les défauts de substitution sont sélectionnés parmi les 56 premiers éléments du tableau périodique (à l'exclusion du diagramme des gaz rares), comme illustré dans la figure ci-dessous. Ils peuvent être introduits individuellement ou en combinaison pour simuler des scénarios de co-dopage réels et servir de référence pour l'ingénierie des défauts dans de futurs espaces de conception à haute dimensionnalité.
Les chercheurs ont utilisé un système de recommandation d'apprentissage automatique pour guider la sélection des dopants : chaque structure a été analysée afin d'identifier les dopants candidats de type n et p. Le cristal initial a ensuite été étendu en une supercellule contenant 433 à 500 atomes, avec des dimensions ajustées de manière adaptative pour garantir une limite inférieure d'environ 0,2% pour la faible concentration de dopage. Les dopants ont été insérés dans le réseau cristallin initial, et chaque structure dopée a subi une relaxation structurale jusqu'à ce que toutes les forces atomiques convergent en dessous de 0,01 eV/Å. Après relaxation structurale, la densité d'états projetée (PDoS) a été calculée par la méthode des déplacements finis pour évaluer les propriétés vibrationnelles, constituant ainsi le spectre d'entrée pour DefectNet.
* La relaxation structurale désigne le processus par lequel l'agencement atomique au sein d'une substance vitreuse se transforme progressivement en une structure plus stable au fil du temps ou lors d'un recuit.
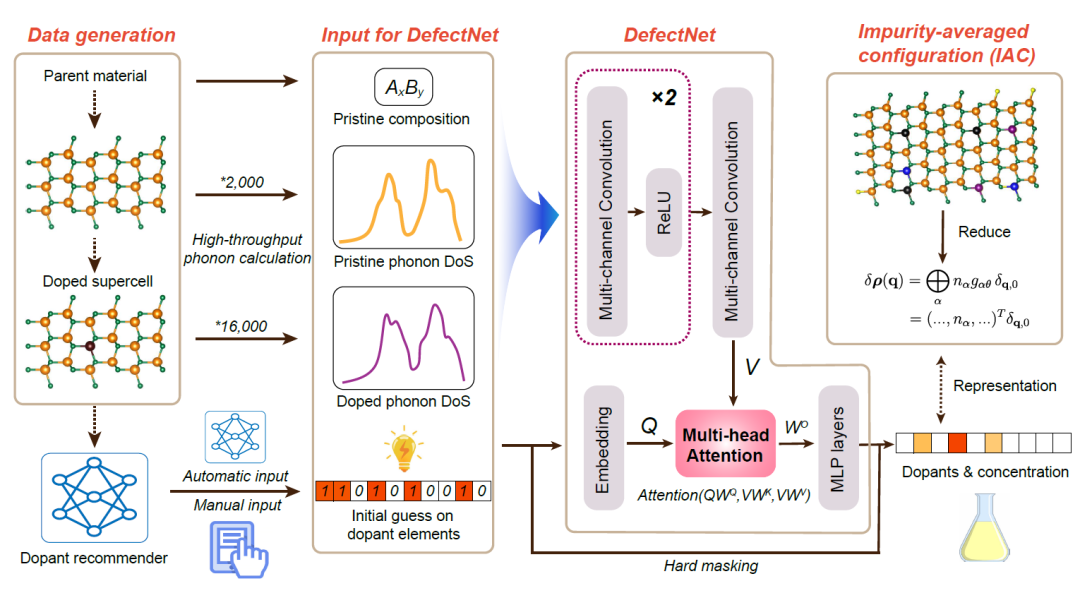
DefectNet : Identifie de manière non destructive les types chimiques et les concentrations de défauts ponctuels directement à partir du PDoS.
DefectNet est utilisé pour prédire les éléments chimiques et les concentrations de défauts à partir des spectres de phonons. Son architecture globale se divise en quatre modules.Le processus est illustré dans la figure ci-dessous. L'objectif du modèle est d'identifier directement le type chimique et la concentration des défauts ponctuels à partir de la densité d'états photoélectroniques (PDoS), sans dommage.
Génération de données
Après avoir constitué l'ensemble de données, les chercheurs ont utilisé le modèle MLIP de base MACE-MP-0 sous forme de phonons figés pour effectuer des calculs de relaxation structurale et de phonons à haut débit sur des cristaux parfaits et des supercellules dopées. Afin de simuler la résolution expérimentale, les courbes PDoS calculées ont également été lissées par un filtre gaussien.
Entrée DefectNet
Le modèle reçoit quatre types d'entrées : la composition du cristal parfait initial, sa densité d'états projetée (PDoS), celle du système dopé et une estimation initiale de la chimie des défauts possibles. Cette estimation initiale peut provenir de l'intuition humaine ou de connaissances existantes, ou être générée automatiquement par un système de recommandation de défauts basé sur l'apprentissage automatique, un modèle probabiliste utilisé pour prédire l'élément dopant le plus probable. Bien que les données d'entraînement soient simulées, ce cadre de travail peut être affiné pour s'adapter à des données spectroscopiques expérimentales (telles que les données de diffusion inélastique de neutrons (INS)).
Architecture du modèle
Le modèle DefectNet est implémenté en PyTorch et adopte une architecture modulaire, composée de quatre parties principales :
* Encodeur spectral basé sur des convolutions 1D :Les données d'entrée sont constituées de trois signaux unidimensionnels de longueur 100 : la densité d'états projetée (PDoS) du matériau non dopé, la PDoS du matériau dopé et le vecteur de composition du cristal hôte. Ces informations sont concaténées en une entrée à trois canaux, et des caractéristiques sont extraites par un réseau de neurones convolutif unidimensionnel pour former finalement 100 « jetons spectraux », chacun étant un vecteur de dimension 128.
* Module d'intégration de dopants :Les estimations initiales des dopants potentiels sont fournies sous forme de vecteurs binaires à 56 dimensions indiquant les éléments dopants envisagés pour un échantillon donné. Ces vecteurs binaires sont projetés, via une couche entièrement connectée, sur le même espace latent à 128 dimensions afin d'obtenir un vecteur d'intégration unique du dopant. Ce vecteur d'intégration sert de requête globale pour le mécanisme d'attention, permettant au modèle d'ajuster l'interprétation de ses caractéristiques spectrales en fonction des contextes de dopage spécifiques.
* Mécanisme d'attention multi-têtes :Pour modéliser l'interaction entre les candidats dopants et les caractéristiques vibrationnelles, DefectNet utilise un mécanisme d'auto-attention multi-têtes, où l'intégration du dopage sert de requête Q et les caractéristiques spectrales V de matrice clé-valeur. Le mécanisme d'attention suit la formule standard du produit scalaire.
* Module à dopant masqué :Le modèle prédit les concentrations de 56 éléments dopants, mais grâce à un mécanisme de masquage strict, seuls les éléments candidats peuvent avoir des valeurs non nulles, et la fonction de perte n'est calculée que pour ces éléments. Ceci présente trois avantages : premièrement, cela améliore la stabilité de l'apprentissage ; deuxièmement, cela évite les interférences des catégories non pertinentes ; et troisièmement, cela assure la cohérence avec les connaissances physiques a priori.
Sortir
Les caractéristiques finales sont masquées de manière stricte en fonction des estimations initiales des défauts, en supprimant les concentrations de dopants absentes de l'ensemble initial. Ce mécanisme garantit que les concentrations de défauts prédites par DefectNet restent confinées à l'ensemble des défauts initialement supposés ; par conséquent, si les estimations initiales sont manquantes ou incomplètes, le modèle risque de ne pas pouvoir identifier certains dopants.
DefectNet peut résoudre 6 défauts de substitution coexistants.
Pour évaluer les capacités de DefectNet, les chercheurs ont conçu une série d'expériences, et les résultats ont montré que :DefectNet peut résoudre jusqu'à six défauts de substitution coexistants à des concentrations aussi faibles que 0,2%, et peut traiter les données PDoS sans nécessiter d'informations détaillées sur la structure atomique.
Application de DefectNet à la prédiction du type et de la concentration des défauts
Les chercheurs ont d'abord testé des semi-conducteurs binaires (SiC, AlAs) et ternaires (AgGaS₂, InCuSe₂) typiques sur un PDoS simulé.
Le SiC et l'AlAs sont très prisés dans les dispositifs électroniques de puissance et la conception d'hétérostructures en raison de leurs larges bandes interdites respectives. La figure ci-dessous présente la densité d'états projetée (PDoS) des cristaux parfaits et dopés, ainsi que les concentrations de dopage prédites et mesurées. Même à de faibles niveaux de dopage (environ 1%), DefectNet parvient à détecter des variations vibrationnelles infimes et à déterminer avec précision la concentration de dopage.
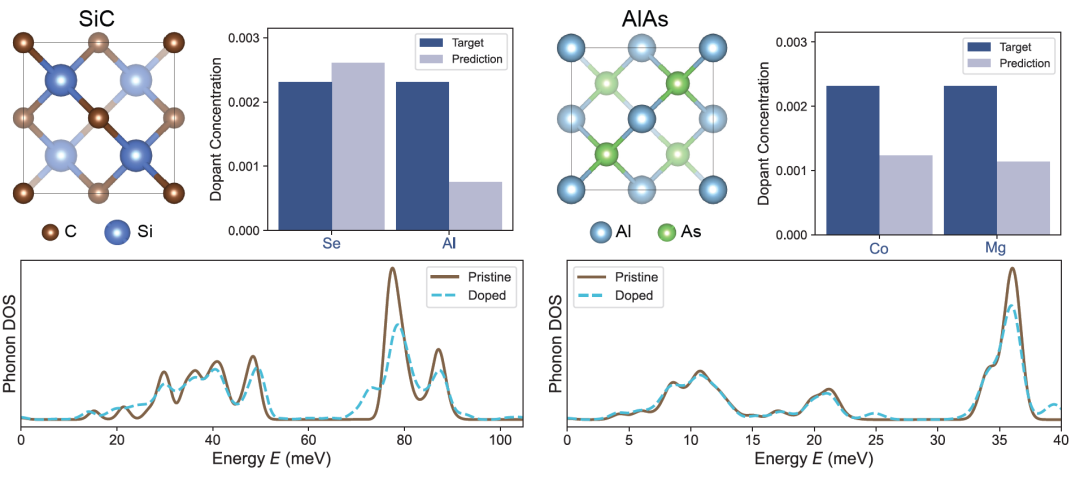
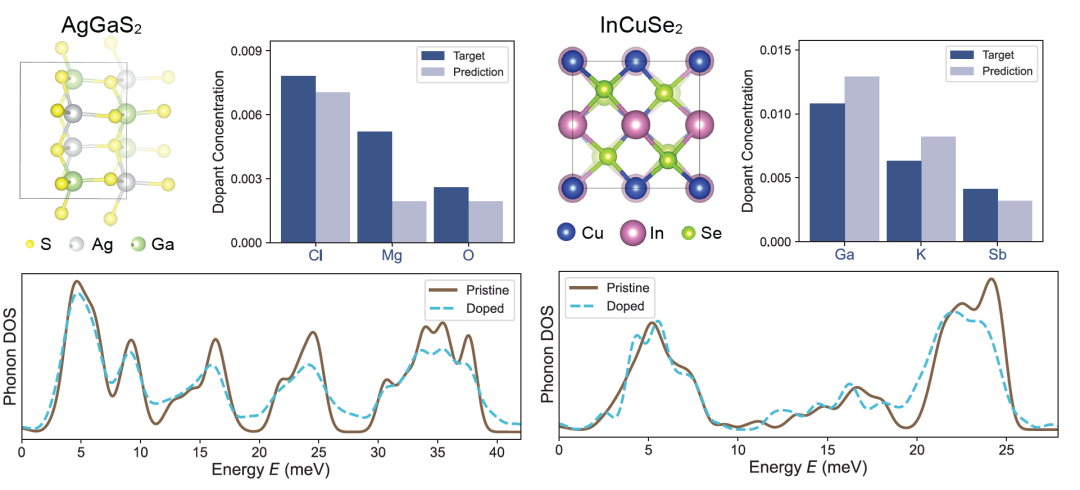
Les chercheurs ont ensuite appliqué DefectNet aux semi-conducteurs ternaires AgGaS₂ et InCuSe₂, plus complexes chimiquement, et les résultats sont présentés dans la figure ci-dessous. AgGaS₂ est utilisé en optique non linéaire infrarouge, tandis qu'InCuSe₂ est un matériau prometteur pour les cellules photovoltaïques en couches minces. Ces matériaux contiennent de multiples sites atomiques non équivalents et divers modes de vibration.Cependant, DefectNet peut toujours suivre les changements de PDoS et déduire la concentration de dopage, démontrant ainsi sa robustesse dans les structures complexes et les systèmes chimiques.
Pour évaluer plus précisément sa capacité de généralisation, les chercheurs ont testé DefectNet sur un jeu de données complet de défauts contenant divers dopants coexistants, y compris des défauts d'« interférence » présents dans les données d'entrée mais inexistants en réalité. La figure ci-dessous présente les résultats en comparant les concentrations de défauts prédites (points colorés) aux valeurs réelles (points noirs), regroupées par quartiles de l'erreur quadratique moyenne (EQM) :
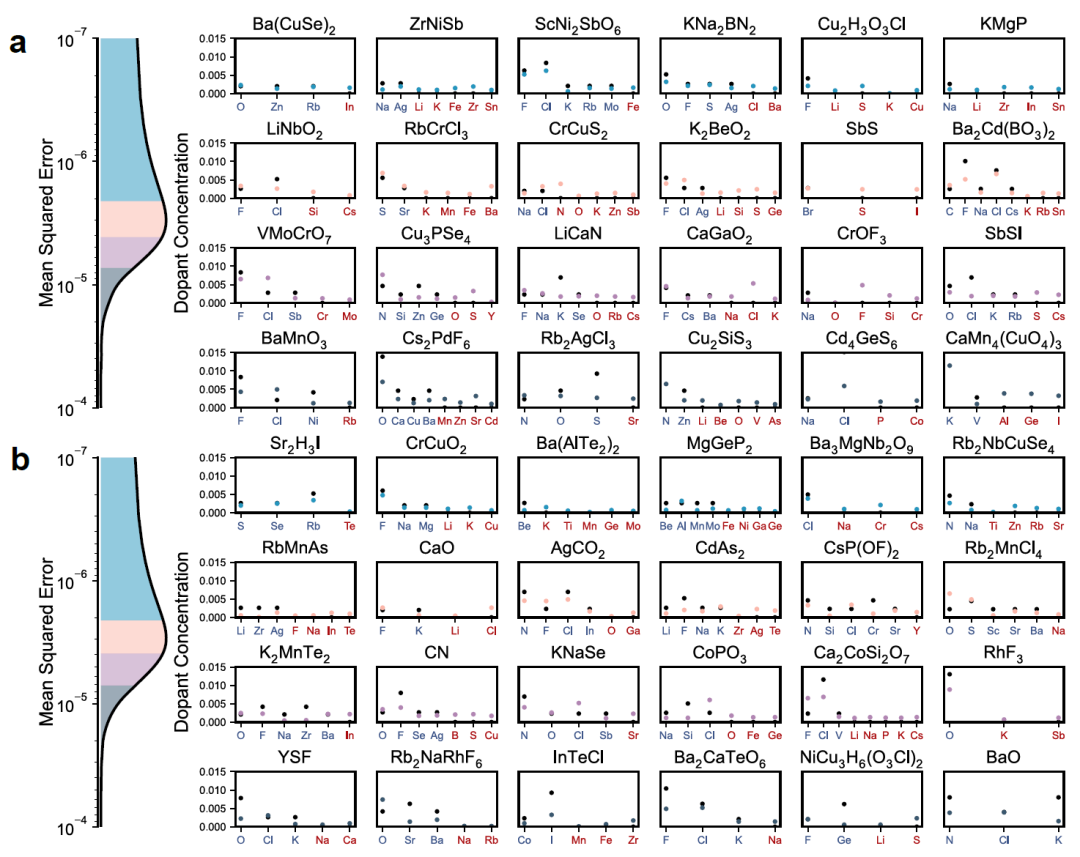
Distribution (Figure a ci-dessus) : Le cristal parent est apparu dans les données d'entraînement, mais ses informations sur les défauts sont inconnues ;DefectNet présente une grande fidélité pour une vaste gamme de types et de concentrations de défauts. Même en présence de défauts parasites dans les données d'entrée, le modèle parvient à identifier le dopage authentique et à éliminer les interférences.
Scénario hors distribution (Figure b ci-dessus) : Le cristal parent n'est pas apparu pendant l'entraînement, ce qui a entraîné une légère diminution de la précision de la prédiction.Cependant, DefectNet est toujours capable de capturer les principales caractéristiques du dopage et d'attribuer des concentrations quasi nulles à la plupart des défauts interférents, démontrant ainsi une bonne capacité de généralisation.
Optimisation de DefectNet à partir de données expérimentales
Pour vérifier l'intérêt pratique de DefectNet, les chercheurs l'ont optimisé et testé sur des données expérimentales. Prenant l'alliage thermoélectrique SiGe comme exemple, ils ont constitué un ensemble d'entraînement composé de 100 supercellules de silicium amorphe. Ces supercellules ont été échantillonnées par simulations de trempe à partir de la base de données Si-GAP-18, couvrant divers états structuraux, du quasi-cristallin à basse énergie au fortement désordonné. Les résultats sont présentés dans la figure ci-dessous :
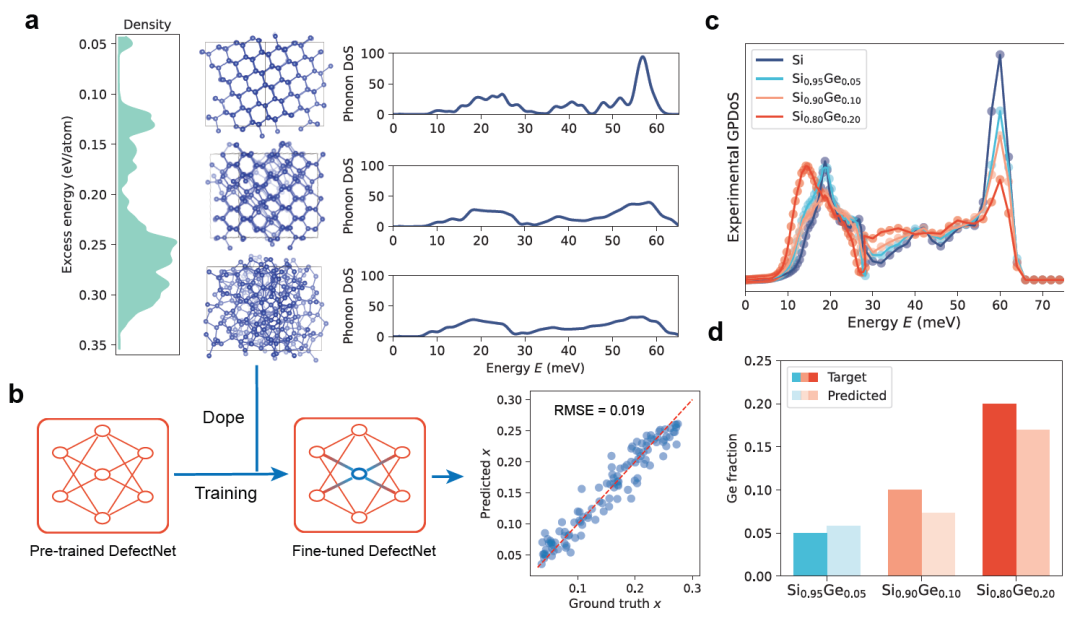
À mesure que le désordre augmente, la courbe PDoS s'élargit et le pic caractéristique des phonons optiques à 60 meV est atténué (figure a ci-dessus), fournissant ainsi un matériau de base pour l'ajustement précis du modèle. Par la suite, les chercheurs ont simulé des alliages SiGe en substituant aléatoirement du Ge dans des supercellules de Si amorphe, couvrant différents niveaux de dopage et différents degrés de désordre, de 0% à 25%.
Après les derniers réglages,DefectNet a atteint une erreur quadratique moyenne (RMSE) de 0,019 sur l'ensemble de test, démontrant une forte performance prédictive (figure b ci-dessus).Le modèle affiné a ensuite été appliqué aux données expérimentales GPDoS de l'alliage Si₁₋ₓGeₓ (x = 5%, 10%, 20%, figure c ci-dessus). DefectNet a prédit des concentrations en Ge de 7%, 13% et 22%, respectivement, ce qui correspond parfaitement à la tendance expérimentale (figure d ci-dessus).
Compte tenu de la difficulté inhérente à la quantification précise des défauts dans les matériaux amorphes, ce résultat démontre la forte capacité prédictive de DefectNet pour les données expérimentales. Pour le supraconducteur multibande dopé à l'aluminium MgB₂, le modèle DefectNet, finement paramétré, peut reproduire les tendances expérimentales jusqu'à une concentration de dopage de 251 TP⁻³T.
Conclusion : Les perspectives sont vastes, mais les défis sont nombreux.
Malgré les perspectives prometteuses de ce modèle, plusieurs défis subsistent quant à son application. Par exemple, à des concentrations de défauts extrêmement faibles, les caractéristiques vibrationnelles sont faibles et facilement masquées par le bruit, ce qui diminue la sensibilité du modèle ; la version actuelle se limite au dopage substitutionnel, et son extension à divers types de défauts ponctuels (tels que les interstitiels, les lacunes, les paires de Frenkel ou les amas de défauts) élargirait considérablement son champ d’application ; bien que les données de simulation présentent une forte capacité de généralisation, un ajustement précis sur les données expérimentales demeure indispensable, et l’obtention d’un modèle directement applicable aux spectres expérimentaux originaux sans réentraînement reste un objectif à long terme.
Pour l'avenir, DefectNet représente une avancée majeure vers un paradigme unifié et fondé sur les données en science des défauts. Son architecture est intrinsèquement compatible avec les entrées spectrales multimodales et ouvre la voie à la conception inverse de matériaux présentant des caractéristiques de défauts spécifiques. En combinant des représentations physiques, des simulations à haut débit, un apprentissage évolutif et un ajustement fin expérimental, DefectNet offre une solution pour l'ingénierie automatisée, interprétable et non destructive des défauts dans des matériaux réels d'une complexité maximale.
Références :
1.https://news.mit.edu/2026/mit-researchers-use-ai-uncover-atomic-defects-materials-0330
2.https://arxiv.org/abs/2506.00725








