Command Palette
Search for a command to run...
Conception De Novo, Pilotée Par l'IA, De Diverses Protéines De Liaison À De Petites Molécules : Une Équipe Sud-coréenne a Découvert Une Protéine Capable De Reconnaître Sélectivement Les Hormones Du stress.

Dans les domaines des sciences de la vie et de la biologie synthétique, la conception de protéines de liaison aux petites molécules présentant à la fois une forte affinité et une grande spécificité a toujours constitué un défi majeur pour la réalisation de biocapteurs et de commutateurs moléculaires. Par le passé, cette approche reposait principalement sur le criblage et la modification de protéines naturelles, ou sur des modélisations physiques basées sur des structures protéiques existantes, ce qui a toujours limité sa polyvalence et son extensibilité.
Compte tenu de cela,Une équipe de recherche du Département des sciences biologiques de l'Institut supérieur coréen des sciences et technologies (KAIST) a utilisé des méthodes de génération de structures protéiques et de conception de séquences basées sur l'apprentissage profond pour concevoir de novo diverses protéines de liaison à de petites molécules, en utilisant le repli de type NTF2 comme « squelette universel » central.De plus, ils ont transformé ce procédé en un capteur similaire à la dimérisation chimiquement induite (CID). Les chercheurs ont conçu avec succès une protéine capable de reconnaître sélectivement le cortisol, l'hormone du stress, et ont développé un biocapteur d'intelligence artificielle basé sur cette protéine. Ce travail dépasse le cadre de la simple conception de protéines, ouvrant la voie à une technologie de capteurs concrètement mesurable et résolvant le problème persistant de la reconnaissance de petites molécules dans le domaine de la conception de protéines.
Les résultats de cette recherche, intitulée « Liaison et détection de petites molécules avec une famille de protéines conçues à cet effet », ont été publiés dans Nature Communications.
Points saillants de la recherche :
* Utiliser l'intelligence artificielle pour concevoir des protéines à partir de zéro (de novo) et les appliquer à des biocapteurs fonctionnels.
Les méthodes traditionnelles consistent principalement à trouver des protéines naturelles ou à modifier certaines fonctions, tandis que cette étude utilise une conception basée sur l'intelligence artificielle pour « personnaliser » les protéines avec les fonctions souhaitées.
* Les résultats de cette recherche peuvent être largement appliqués dans des domaines tels que le diagnostic des maladies, le développement de nouveaux médicaments et la surveillance environnementale.

Adresse du document :
https://www.nature.com/articles/s41467-026-70953-8
Autres articles sur les frontières de l'IA :
https://hyper.ai/papers
Jeu de données : Construction de l’infrastructure NTF2
Pour atteindre les objectifs de conception,Les chercheurs ont d'abord généré un ensemble de structures NTF2 (ensemble 1 : 1 615 squelettes) à l'aide d'une méthode d'« hallucination » au niveau de la famille, puis ont utilisé ProteinMPNN pour redessiner les séquences de ces squelettes.L'algorithme AlphaFold a été utilisé pour sélectionner les protéines capables de se replier selon les structures conçues (ensemble 2 : 3 230 squelettes). De plus, la paramétrisation Rosetta a permis de générer la structure du squelette, et ProteinMPNN a été utilisé pour la conception de la séquence, suivi d'AlphaFold pour la validation de la structure (ensemble 3 : 6 838 squelettes), comme illustré dans la figure ci-dessous :
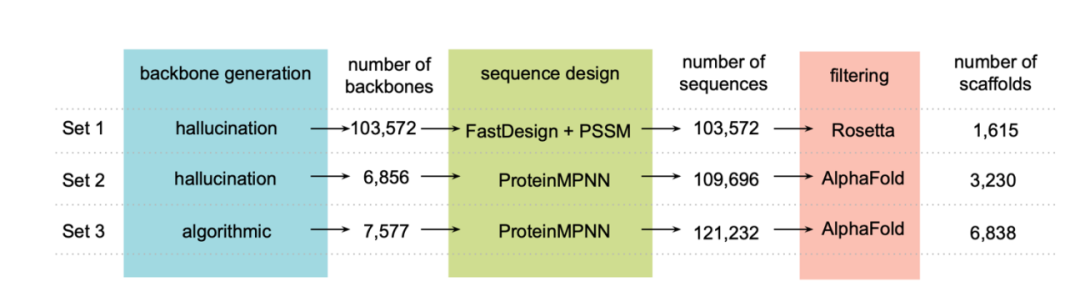
Finalement, après sélection, les chercheurs ont également obtenu les oligonucléotides codants pour la caractérisation expérimentale, notamment : 630 protéines de liaison HCY, 1 661 protéines de liaison ROC, 16 276 protéines de liaison WRF, 9 024 protéines de liaison APX, 19 390 protéines de liaison IRI et 7 573 protéines de liaison OHP.
Conception de la famille de protéines NTF2 avec des géométries de poche diversifiées
Le repliement NTF2 est constitué de trois hélices α et d'un feuillet β incurvé à 6 brins. Ces structures forment ensemble la grande poche de connexion interne caractéristique de cette famille de repliements, comme illustré dans la figure ci-dessous :
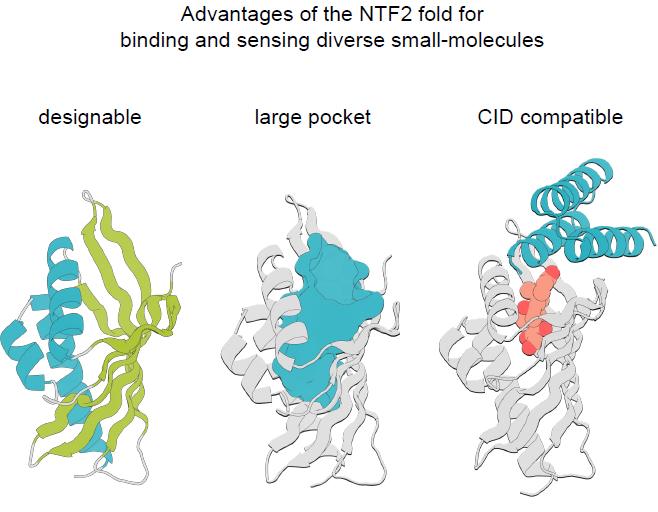
La diversité de ce repliement dans la nature provient principalement des anneaux longs et irréguliers et de la structure quaternaire unique, qui influencent tous deux la géométrie et la fonction de la poche combinée.L'objectif de cette étude est de concevoir une famille de protéines NTF2 avec des géométries de poche diverses pour accueillir une large gamme de petites molécules, tout en minimisant les régions de boucle afin de maintenir leur modularité et leur capacité de conception.Le processus de conception global est illustré dans le diagramme suivant :
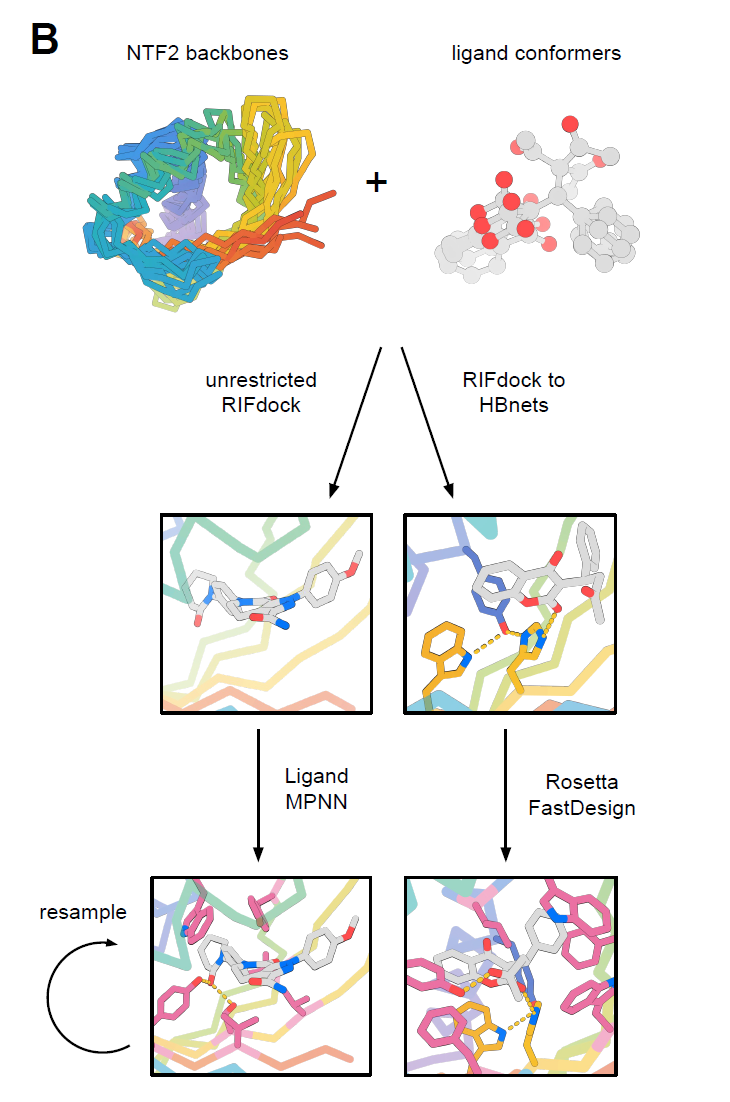
Après avoir obtenu plus de 10 000 protéines NTF2 conçues avec diverses géométries de poches, les chercheurs ont utilisé RIFdock pour insérer six petites molécules, différant par leurs propriétés chimiques et leur structure, dans les poches centrales de ces squelettes. Ces petites molécules comprenaient l’hormone du stress cortisol (HCY), l’anticoagulant warfarine (WRF), le myorelaxant bromure de rocuronium (ROC), l’anticoagulant apixaban (APX), la molécule antitumorale active SN-38 (IRI) dérivée de l’anticancéreux irinotécan, et l’hormone 17-α-hydroxyprogestérone (OHP).
La construction d'interfaces polaires représente un défi majeur dans la conception des protéines, notamment pour les protéines de liaison aux petites molécules. Cela nécessite l'introduction de résidus polaires dans la poche interne afin d'interagir avec les groupements fonctionnels polaires des ligands, tout en préservant la stabilité globale de la protéine. Pour y parvenir, les chercheurs ont mis en œuvre deux stratégies :
Méthode 1 (RIFdock vers HBNets)
Les chercheurs ont intégré les séquences HCY, WRF, ROC, APX et IRI au squelette peptidique et ont exigé au moins une interaction protéine-petite molécule via les résidus HBNet. Ils ont ensuite optimisé la structure à l'aide de Rosetta, un outil de conception guidé par des séquences naturelles. Ce dernier utilisait une matrice de score spécifique à chaque position, dérivée des protéines de la famille NTF2, pour orienter la conception de la séquence.
Méthode 2 (RIFdock sans restriction)
Les ligands OHP, APX et IRI ont été intégrés aux squelettes des ensembles 2 et 3 à l'aide de RIFdock sans contrainte, et la conception des séquences a été réalisée avec LigandMPNN. LigandMPNN est une variante de ProteinMPNN spécifiquement entraînée sur des données de complexes protéine-petite molécule, permettant ainsi de prendre explicitement en compte la présence du ligand lors de la conception.
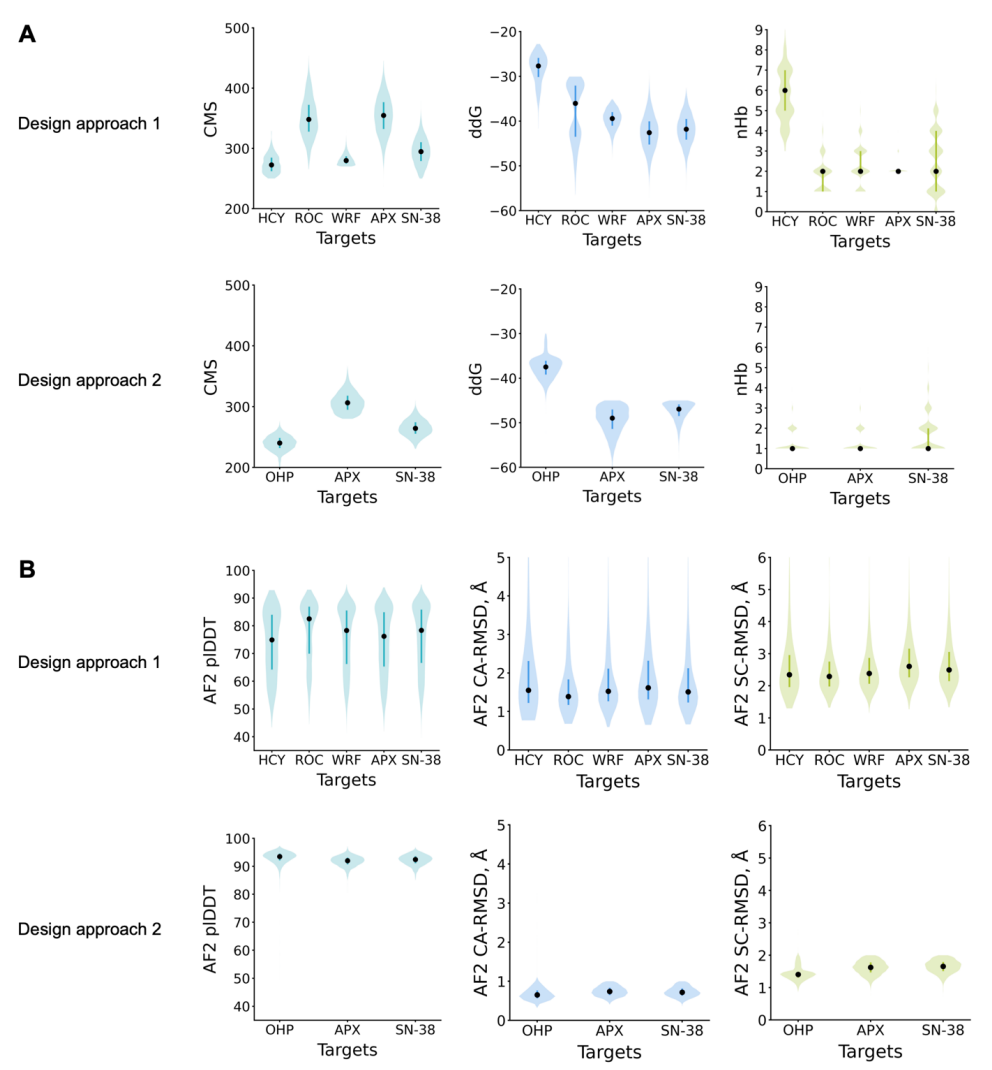
Lors du criblage des résultats de conception, les chercheurs ont utilisé Rosetta pour calculer le nombre de liaisons hydrogène entre la protéine et le ligand, l'énergie de liaison (ddG) et la surface moléculaire de contact (CMS) ; pour la méthode 2, ils ont également combiné les résultats de prédiction AlphaFold à séquence unique pour cribler les conceptions qui pouvaient simultanément reproduire la structure de repliement cible et le site de liaison (voir figure ci-dessous).
Présentation des résultats : Les protéines de liaison aux petites molécules basées sur NTF2 peuvent être utilisées dans les biocapteurs
Les chercheurs ont conçu une série d'expériences pour vérifier l'efficacité de la stratégie de conception proposée dans cette étude :
Conception et caractérisation structurale des protéines de liaison
Afin de vérifier la précision des protéines de liaison aux petites molécules conçues, les structures cristallines de deux complexes protéine-ligand ont été résolues : la protéine de liaison au cortisol hcy129 et la protéine de liaison à l’apixaban apx1049. Plus précisément, la surface de hcy129 a été modifiée à l’aide de ProteinMPNN pour améliorer sa cristallinité, permettant ainsi d’obtenir sa structure à haute résolution (1,5 Å) en complexe avec le cortisol. L’alignement structural a montré que le repliement global était très conforme au modèle de conception, avec un RMSD Cα de 1,1 Å (Figure A ci-dessous). Les résidus clés des liaisons hydrogène et les conformations du ligand correspondaient également précisément (Figure B ci-dessous), indiquant que le réseau de liaisons hydrogène pré-construit (HBNet) a permis une conception précise des interactions polaires.
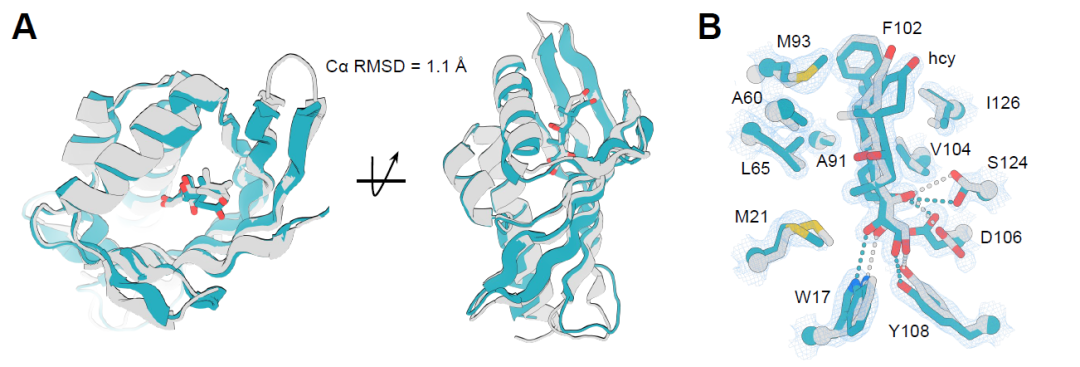
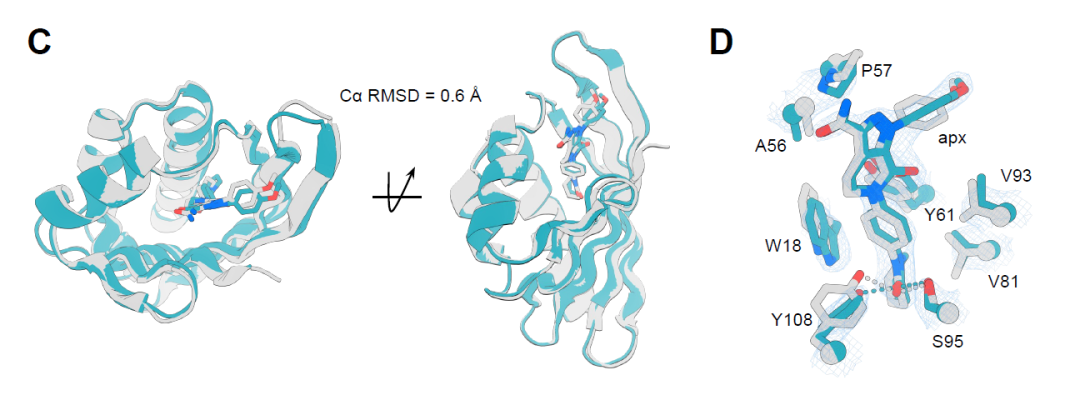
En revanche, la structure cristalline du complexe apx1049-apixaban présente une résolution de 2,1 Å, témoignant d'une meilleure concordance avec le modèle théorique, et un écart quadratique moyen (RMSD) des atomes Cα de seulement 0,6 Å sur une plage de 113 résidus (Figure C ci-dessous). Ses interactions protéine-ligand reproduisent presque parfaitement le modèle théorique, notamment les liaisons hydrogène clés et les interactions π–π entre les résidus aromatiques (Figure D ci-dessous), stabilisant ainsi la conformation du ligand et formant une poche de liaison parfaitement complémentaire. Ces résultats démontrent que cette stratégie de conception permet une construction d'interface protéine-ligand de haute précision à l'échelle atomique.
Conception d'une évaluation de spécificité pour les protéines de liaison
Pour évaluer la spécificité des protéines conçues, l'étude a testé systématiquement six protéines de liaison avec six ligands, en utilisant l'albumine, qui possède une capacité de liaison non spécifique, comme témoin. Les résultats ont montré que les protéines à haute affinité telles que hcy129.1, iri807.1 et apx1049 présentaient une bonne spécificité de liaison à leurs cibles respectives, tandis que l'albumine ne présentait quasiment aucune liaison à la plupart des ligands, validant ainsi l'efficacité de la stratégie de conception.
De plus, dans le système de warfarine (WRF), l'affinité de liaison de l'albumine (KD d'environ 5,0 μM) à celle de la protéine conçue wrf1071 (KD d'environ 1,1 μM), indiquant que la liaison non spécifique à des ligands hautement hydrophobes reste difficile.
Globalement, cette méthode a permis d'atteindre un certain degré de spécificité élevée en matière de reconnaissance, mais il reste encore des possibilités d'optimisation pour distinguer les molécules structurellement similaires et améliorer la sélectivité pour les ligands hydrophobes.
Construction de biocapteurs (Conception et caractérisation d'hétérodimères induits par le cortisol)
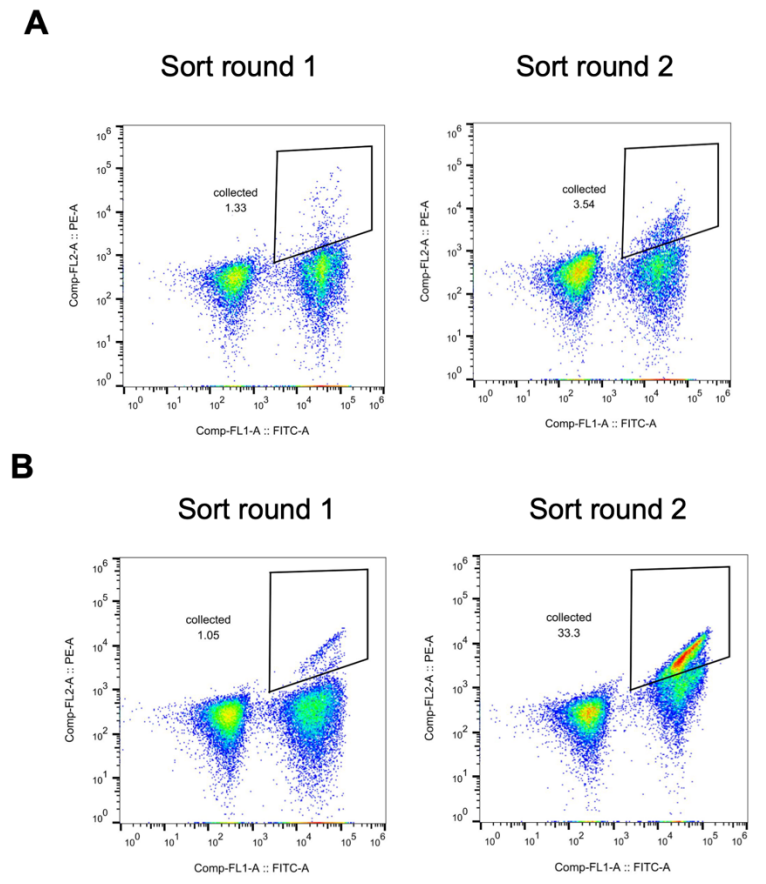
Le cortisol est généralement présent dans les échantillons physiologiques à de faibles concentrations nanomolaires, mais des taux plasmatiques supérieurs à 38 nM peuvent servir de critère diagnostique pour des maladies telles que le syndrome de Cushing. Afin d'améliorer l'affinité de liaison de l'hcy129 pour le cortisol en vue de sa biodétection, des chercheurs ont construit une banque de mutants combinatoires à partir de mutations favorables identifiées lors d'expériences de saturation de mutations ponctuelles (SSM). Le criblage a été réalisé par la technique du « leishing display », et une augmentation significative de l'affinité de liaison a été observée, comme illustré dans la figure ci-dessous.
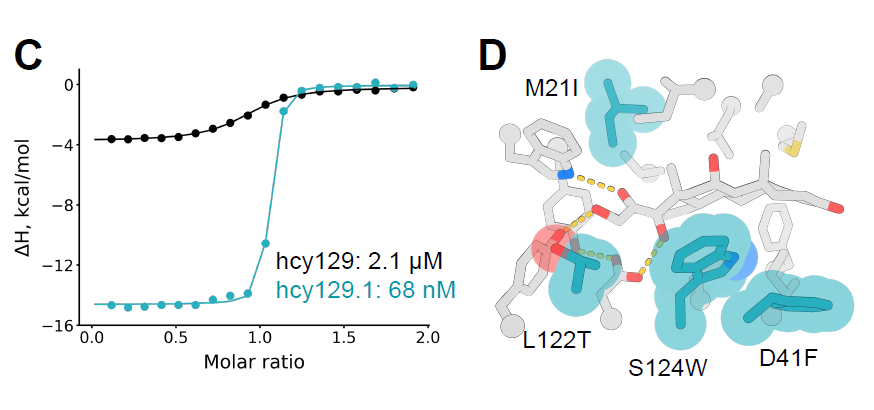
Par la suite, les chercheurs ont sélectionné la variante optimale dans la bibliothèque, l'ont exprimée dans E. coli et l'ont caractérisée par calorimétrie de titration isotherme (ITC). Le KD de cette variante hcy129.1 était de 68 nM, soit 31 fois supérieur à celui de la variante originale (Figure C ci-dessous) ; l'analyse structurale a montré que l'affinité accrue provenait principalement d'une interaction hydrophobe plus forte avec le cortisol (Figure D ci-dessous).
S’appuyant sur ces travaux, l’étude a ensuite conçu un système hétérodimère dépendant du cortisol. En modifiant la structure de l’hcy129.1 et en y introduisant un petit squelette protéique, une conception et un criblage informatiques ont été réalisés à l’aide de méthodes telles que RIFdock, Rosetta et ProteinMPNN. Finalement, une petite protéine, miniH11, a été obtenue, capable de former un complexe ternaire avec l’hcy129.1 et le cortisol.
Des expériences ont montré que le système forme un complexe stable uniquement en présence de cortisol. De plus, le système a été fusionné à un système de luciférase NanoBiT pour permettre la détection du cortisol, avec une CE50 d'environ 72 nM (Figure H ci-dessous), cohérente avec l'affinité de liaison, validant ainsi l'efficacité du dispositif. Parallèlement, l'affinité du système diminue significativement en l'absence de cortisol, indiquant que le processus de dimérisation est fortement dépendant du ligand.
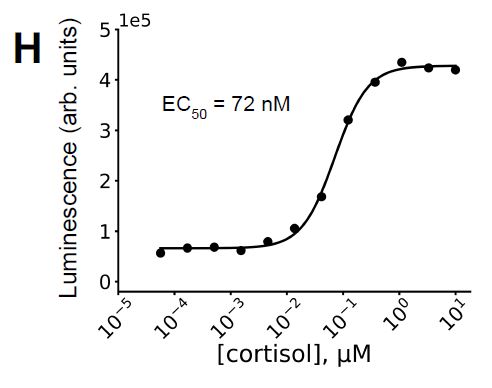
Dans le système équimolaire (200 nM) hcy129.1_CID-SmBiT et miniH11-LgBiT, les courbes de réponse de luminescence dépendantes du cortisol...
Dans l'ensemble,Ce travail démontre que les protéines de liaison aux petites molécules basées sur NTF2 peuvent être transformées en biocapteurs fonctionnels.
Conclusion
Globalement, cette étude propose une nouvelle voie pour la conception de novo de protéines de liaison aux petites molécules : en utilisant des modèles d’intelligence artificielle pour caractériser précisément les interactions protéine-ligand au niveau atomique, elle permet de passer de la « découverte ou de la modification de protéines naturelles » à la « personnalisation de protéines fonctionnelles à la demande » et effectue une validation efficace au niveau expérimental.
Cette avancée majeure représente non seulement un bond en avant dans la conception des protéines, mais élargit aussi considérablement leur champ d'application : de la détection précise de biomarqueurs pour le diagnostic précoce des maladies à la reconnaissance moléculaire ciblée pour le développement de nouveaux médicaments, en passant par la perception en temps réel des polluants pour la surveillance environnementale. À mesure que cette technologie se perfectionne, des biocapteurs personnalisés, hautement spécifiques et programmables devraient devenir un lien essentiel entre les sciences de la vie et leurs applications concrètes.
Références :
https://www.nature.com/articles/s41467-026-70953-8
https://phys.org/news/2026-04-ai-proteins-built-specific-compounds.html








