Command Palette
Search for a command to run...
Dépasser Les Limites De l'intégration Multimodale Traditionnelle ! Le MIT Propose Le Cadre APOLLO, Qui Permet Une Séparation Claire Entre Les Informations Partagées Par Les Cellules Et Les Informations Spécifiques À Chaque cellule.
En biologie unicellulaire, le développement rapide des technologies de mesure repousse sans cesse les frontières de l'exploration scientifique. Des avancées majeures dans des domaines tels que l'imagerie multiplex, le séquençage du transcriptome unicellulaire (scRNA-seq), le séquençage de la chromatine ouverte (scATAC-seq) et la détection de l'abondance des protéines ont permis aux chercheurs d'effectuer des observations panoramiques de cellules uniques selon de multiples dimensions, notamment la régulation transcriptionnelle, l'état de la chromatine, l'expression des protéines et la structure morphologique. Ces données multimodales interprètent le code de la vie à différents niveaux, et leur intégration complémentaire offre une opportunité sans précédent de révéler l'hétérogénéité cellulaire et d'explorer les mécanismes des maladies.
Cependant, les méthodes analytiques actuelles présentent encore des limitations importantes lorsqu'il s'agit de traiter des données à haut débit.Les stratégies classiques consistent souvent à analyser chaque mode séparément puis à les comparer, ce qui est non seulement inefficace, mais rend également difficile la saisie des corrélations profondes entre les modes.Une autre approche consiste à intégrer des données multimodales dans un même espace latent grâce à l'apprentissage de représentations, mais elle confond souvent les informations partagées avec les informations spécifiques à chaque modalité, masquant ainsi la contribution unique de chaque dimension à la fonction cellulaire.
Ce problème est particulièrement visible dans l'analyse intégrée des données appariées scATAC-seq et scRNA-seq.Les méthodes traditionnelles granulent souvent grossièrement l'accessibilité de la chromatine au niveau du gène pour la comparer à l'expression génique. Bien que cela simplifie le problème, cette approche peut entraîner la perte d'informations structurales fines au niveau de la chromatine et n'est applicable qu'aux types de données présentant des caractéristiques relativement uniformes. Les méthodes d'intégration plus complexes, telles que les modèles linéaires et les réseaux antagonistes génératifs (GAN), peinent soit à s'adapter aux données non structurées comme l'imagerie, soit à distinguer les informations partagées des informations spécifiques, ne répondant ainsi pas à la demande croissante d'analyse de données multimodales dans les grandes biobanques.
Par conséquent, avec l'évolution continue de la technologie unicellulaire et la croissance rapide du volume de données, la manière d'intégrer efficacement et automatiquement des données multimodales tout en dissociant clairement les informations partagées des informations spécifiques à chaque modalité est devenue un défi fondamental pour la biologie unicellulaire.
Pour relever ce défi, une équipe de recherche conjointe du MIT et de l'ETH Zurich a proposé un cadre de calcul d'apprentissage profond général appelé APOLLO (Autoencoder avec un espace latent partiellement chevauchant appris par optimisation latente).Ce cadre offre une voie technique réalisable pour une analyse plus complète et précise des états cellulaires et de leur logique de régulation en modélisant explicitement les informations partagées et les informations spécifiques à chaque modalité.
Les résultats de cette recherche, intitulée « Partially shared multimodal embedding learns holistic representation of cell state », ont été publiés dans Nature Computational Science.
Points saillants de la recherche :
Cette recherche propose APOLLO, un cadre d'apprentissage profond général capable de découpler automatiquement et explicitement les « informations partagées » des « informations spécifiques à la modalité » dans les données multimodales.
* APOLLO apprend un espace latent partiellement chevauchant en équipant chaque modalité d'un auto-encodeur et en employant une stratégie d'entraînement en deux étapes, identifiant et distinguant ainsi efficacement les signaux biologiques couramment capturés à travers plusieurs modalités.
* APOLLO peut révéler l'association entre les différences de localisation subcellulaire des protéines et la morphologie des différents compartiments cellulaires, étendant ainsi l'analyse des données purement omiques au domaine de la morphologie spatiale.

Adresse du document :
https://www.nature.com/articles/s43588-025-00948-w
Suivez notre compte WeChat officiel et répondez « APOLLO » en arrière-plan pour obtenir le PDF complet.
Jeu de données : Validation complète couvrant le séquençage et l’imagerie
Pour évaluer de manière exhaustive les performances du cadre APOLLO, l'étude a utilisé plusieurs ensembles de données multimodales monocellulaires disponibles publiquement, couvrant à la fois les technologies de séquençage et d'imagerie.
Concernant les données de séquençage,Les chercheurs ont d'abord utilisé des données appariées de transcriptome unicellulaire (scRNA-seq) et d'accessibilité de la chromatine (scATAC-seq) mesurées par la technologie SHARE-seq pour vérifier si APOLLO pouvait automatiquement identifier et distinguer l'activité génique capturée à la fois par le transcriptome et l'accessibilité de la chromatine, ainsi que l'activité génique capturée par une seule modalité de l'une ou l'autre.
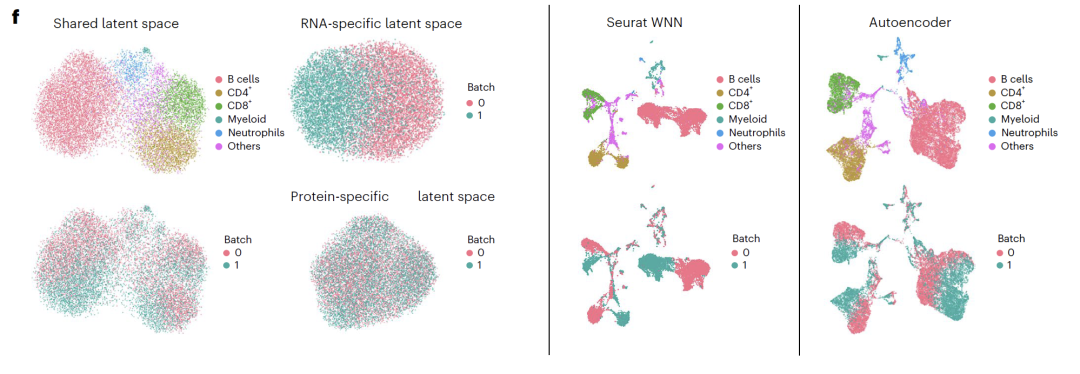
Deuxièmement, les chercheurs ont utilisé des données appariées scRNA-seq et d'abondance de protéines de surface cellulaire obtenues par CITE-seq pour tester plus avant l'applicabilité du modèle aux données de séquençage.L'ensemble de données CITE-seq est dérivé de rates et de ganglions lymphatiques de souris et comprend deux groupes d'échantillons de souris de type sauvage qui ont subi un traitement expérimental indépendant.Elle peut non seulement servir à évaluer la capacité de discrimination des types cellulaires, mais aussi à révéler l'effet de lot expérimental causé par différentes sources de souris.
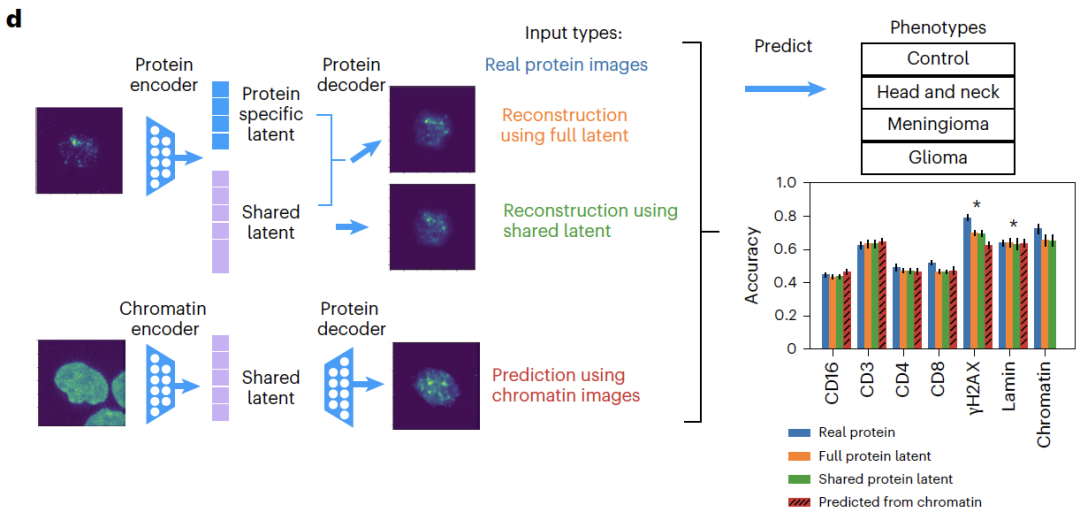
Concernant les données d'imagerie,Des chercheurs ont présenté un jeu de données d'imagerie multiplex de cellules mononucléaires du sang périphérique (PBMC) humaines, comprenant 32 345 cellules provenant de 40 patients et classées en quatre catégories diagnostiques : sujets sains, méningiomes, gliomes et tumeurs de la tête et du cou. Deux séries de données d'imagerie ont été recueillies pour chaque patient, selon différentes combinaisons d'anticorps : la première série utilisait le DAPI pour marquer la chromatine et était combinée à un marquage par anticorps anti-CD4, anti-CD8 et anti-CD16 ; la seconde série utilisait également le marquage au DAPI, mais combiné à un marquage par anticorps anti-lamine, anti-CD3 et anti-γH2AX.
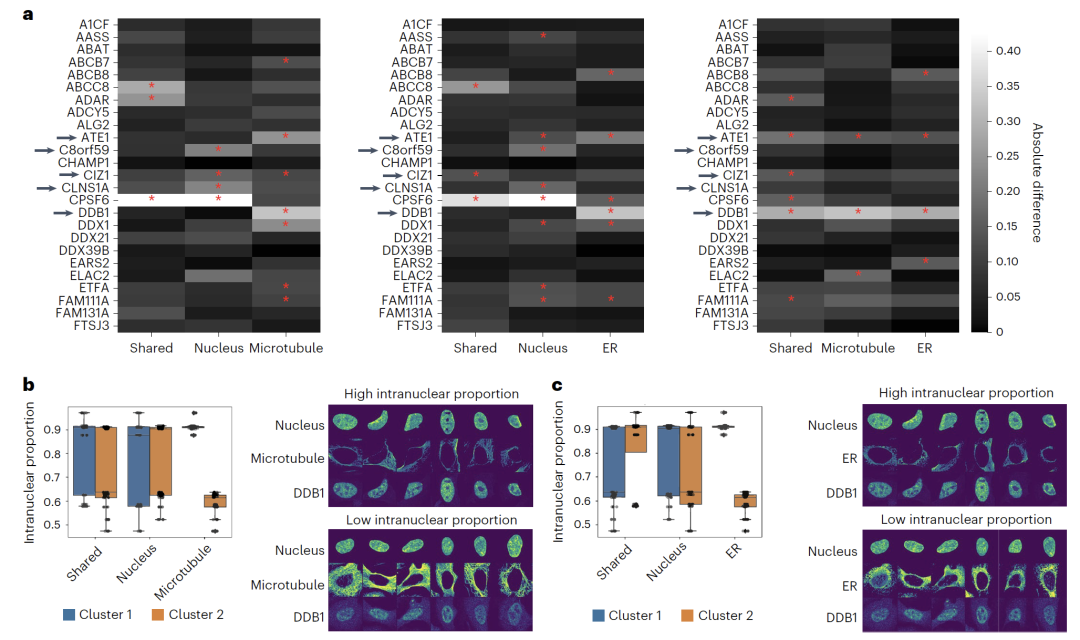
Les tests effectués à l'aide de cet ensemble de données ont révélé que,APOLLO peut identifier les informations sur l'état cellulaire partagées entre les deux modalités dans la structure de la chromatine et la localisation des protéines, ainsi que les caractéristiques morphologiques capturées par une seule modalité.En outre, en combinant des marqueurs de coloration cellulaire supplémentaires tels que les microtubules et le réticulum endoplasmique, l'étude a également utilisé de multiples données d'imagerie de l'Atlas des protéines humaines (HPA) pour démontrer qu'APOLLO peut être utilisé pour révéler l'association entre les différences de localisation subcellulaire des protéines et la morphologie des différents compartiments cellulaires.
Modèle APOLLO : Un auto-encodeur utilisant une stratégie d’optimisation latente
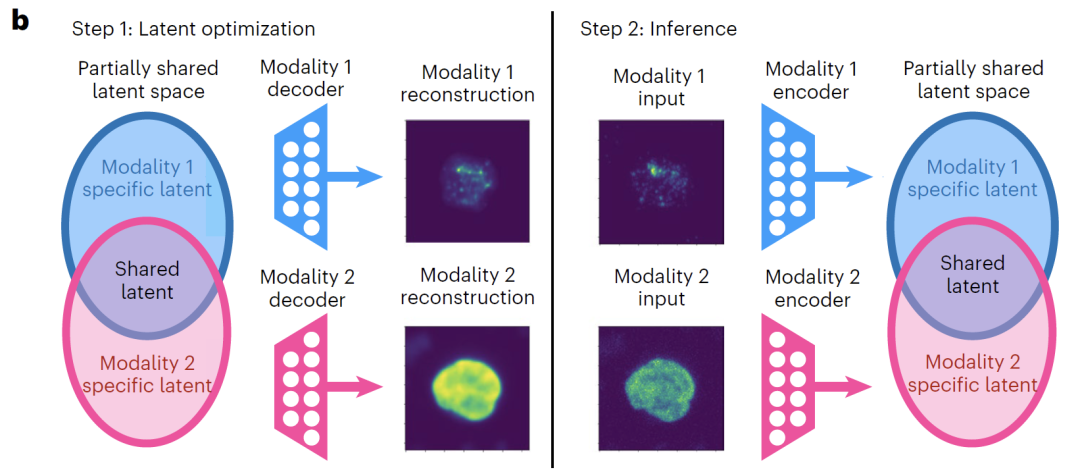
Pour remédier au problème commun aux méthodes d'intégration multimodale existantes, qui confondent informations partagées et informations spécifiques à chaque modalité, cette étude propose le cadre APOLLO. Ce cadre utilise l'optimisation latente pour apprendre un auto-encodeur dans un espace latent partiellement chevauchant, afin d'apprendre automatiquement et de découpler efficacement les informations partagées et spécifiques à chaque modalité. Contrairement aux auto-encodeurs conventionnels qui alignent uniformément toutes les dimensions latentes,APOLLO effectue un alignement intermodal uniquement sur certaines des dimensions potentielles, réservant les dimensions restantes aux informations spécifiques à chaque modalité, réalisant ainsi une séparation claire entre les informations partagées et spécifiques dans la conception du modèle.
En termes d'architecture du modèle,APOLLO est équipé d'un autoencodeur pour chaque modalité de données et peut intégrer des décodeurs supplémentaires selon les besoins de la tâche.L'encodeur et le décodeur utilisent des architectures de réseaux neuronaux adaptées aux modalités spécifiques ; par exemple, des réseaux convolutionnels sont utilisés pour les données d'imagerie, tandis que des réseaux entièrement connectés sont utilisés pour les données d'expression génique, afin de capturer pleinement les caractéristiques de chaque modalité. L'espace latent est explicitement divisé en deux parties : les caractéristiques latentes partagées et les caractéristiques latentes spécifiques à chaque modalité. La dimension de l'espace latent partagé est généralement beaucoup plus grande que celle de l'espace spécifique à chaque modalité afin de garantir une représentation suffisante des informations partagées entre les modalités.
Comme le montre la figure ci-dessous, le processus d'entraînement d'APOLLO se compose de deux étapes :La première étape consiste à entraîner les décodeurs pour chaque modalité, tout en mettant à jour simultanément l'espace latent.L'objectif principal est de permettre au décodeur de reconstruire avec précision les données d'entrée à partir de l'espace latent. Si la tâche requiert un renforcement de la représentation des informations partagées et la réalisation d'une prédiction intermodale, deux décodeurs supplémentaires sont introduits pour mapper l'espace latent partagé sur chaque modalité respectivement, et l'entraînement est finalisé en minimisant la perte de reconstruction.
La deuxième étape consiste à entraîner l'encodeur spécifique à la modalité.Chaque type de données est associé à son espace latent correspondant. En minimisant l'erreur quadratique moyenne, on infère l'intégration dans l'espace latent des échantillons non utilisés pour l'entraînement, ce qui garantit une bonne capacité de généralisation du modèle.
Pour la validation du modèle, l'étude a d'abord testé les performances de découplage d'APOLLO sur cinq ensembles de données simulées avec des structures sous-jacentes réelles connues.Les résultats montrent que le modèle peut maintenir des performances stables quelle que soit la dépendance entre les caractéristiques latentes partagées et spécifiques.Une validation supplémentaire sur des données réelles montre que l'apprentissage explicite par APOLLO du partage partiel d'informations peut non seulement découpler les informations multimodales, mais aussi réaliser des prédictions intermodales précises, telles que la prédiction de protéines non détectées à partir de l'imagerie de la chromatine.
Globalement, APOLLO découple et interprète efficacement les informations partagées et spécifiques à une modalité dans les ensembles de données multimodaux en apprenant des espaces latents partiellement partagés, fournissant un cadre général pour découvrir les mécanismes biologiques.
Au-delà des cadres d'intégration multimodaux traditionnels, une compréhension plus globale des états cellulaires.
Afin d'évaluer de manière exhaustive l'universalité et les principaux avantages du modèle APOLLO, une série d'expériences ont été conçues autour de cinq axes : l'intégration des données de séquençage par paires, l'intégration de l'imagerie de la chromatine et des protéines, la prédiction intermodale, la reconnaissance des caractéristiques morphologiques et l'exploration de la localisation subcellulaire des protéines.
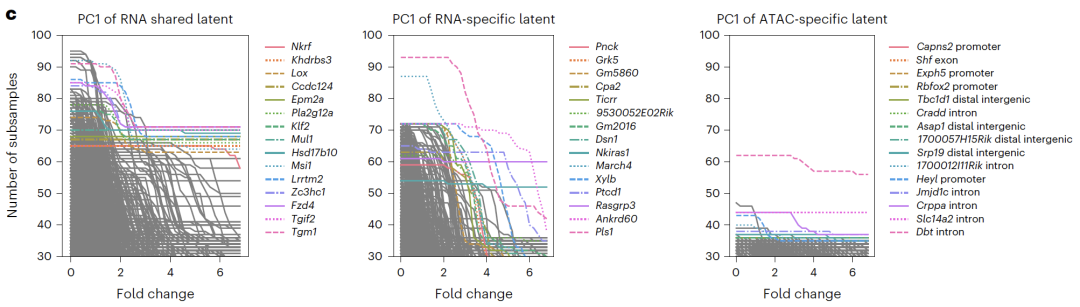
Dans l'intégration des données de séquençage appariéLes expériences SHARE-seq ont montré que l'ajout d'un espace spécifique à une modalité à l'espace partagé peut améliorer considérablement la précision de la classification des types cellulaires, prouvant ainsi que l'espace spécifique peut capturer des informations biologiques non incluses dans l'espace partagé.
L'interprétation spatiale potentielle a révélé que les espaces spécifiques à l'ARN étaient enrichis en gènes liés au cycle cellulaire, les espaces spécifiques à l'ATAC étaient enrichis en régions de chromatine ouverte liées à la régulation transcriptionnelle, et les espaces partagés étaient enrichis en facteurs de transcription et voies de régulation connus, validant ainsi la signification biologique des résultats de découplage. Dans les expériences CITE-seq,APOLLO a réussi à séparer les effets liés au type cellulaire et au lot en un espace commun et un espace spécifique à l'ARN.Les méthodes d'intégration existantes ne permettent pas d'atteindre ce type de découplage, ce qui souligne les avantages uniques du modèle en matière d'intégration des données de séquençage.
Concernant les données d'imagerie,APOLLO peut reconstruire avec précision les images cellulaires de patients n'ayant pas participé à la formation.Dans la tâche intermodale de prédiction des protéines non détectées à partir de la chromatine, APOLLO surpasse significativement les méthodes traditionnelles de remplissage d'images ;La classification phénotypique en aval a montré que la précision de la classification basée sur l'imagerie protéique prédite était similaire à celle de l'imagerie réelle, la protéine CD3 présentant les meilleures performances de prédiction, confirmant ainsi que les résultats de la prédiction peuvent être utilisés efficacement pour la découverte biologique.
Dans les tâches de reconnaissance de caractéristiques morphologiques,L'espace partagé capture principalement les caractéristiques morphologiques de la chromatine (telles que la surface nucléaire et le volume d'hétérochromatine), tandis que les caractéristiques spécifiques aux protéines, comme le nombre de foyers γH2AX, n'existent que dans leurs espaces spécifiques respectifs. Des expériences d'ablation de caractéristiques ont montré que la suppression de cette caractéristique réduisait significativement la précision de la classification phénotypique, validant ainsi la précision du découplage.

Dans l'étude de la localisation subcellulaire des protéinesL'application d'APOLLO aux données d'imagerie cellulaire U2OS a révélé que les différences de localisation des protéines au sein de la cavité nucléaire peuvent être mises en évidence par les caractéristiques de différents compartiments cellulaires. Par exemple, la localisation nucléaire de DDB1 est associée à la morphologie du réticulum endoplasmique et des microtubules, tandis que celle de CLNS1A est uniquement associée à la morphologie nucléaire. Ce résultat indique que…Ce modèle peut être étendu à diverses combinaisons d'imagerie, offrant une nouvelle perspective pour comprendre la relation entre la localisation des protéines et la morphologie cellulaire.
Mise en œuvre de l'intégration de données multimodales monocellulaires
L'intégration de données multimodales unicellulaires est en passe de devenir une orientation technologique essentielle pour l'analyse de l'hétérogénéité cellulaire, la mise en évidence des mécanismes pathologiques et la promotion du développement de la médecine de précision, et a suscité un vif intérêt au sein de la communauté universitaire mondiale.
Par exemple, la technologie scMTR-seq développée par l'équipe de Peter Rugg-Gunn à l'Institut Babraham de l'Université de Cambridge...Pour la première fois, la capture simultanée de six modifications d'histones et de l'ensemble du transcriptome a été réalisée au niveau de la cellule unique.Ils ont surmonté un obstacle technique qui persistait depuis une décennie dans le domaine de la recherche en épigénétique.
Titre de l'article :
Profilage combinatoire de multiples modifications d'histones et du transcriptome dans des cellules uniques par scMTR-seq
Lien vers l'article :
https://www.science.org/doi/10.1126/sciadv.adu3308
Le cadre CellFuse proposé par l'équipe de recherche de l'université de Stanford construit un espace d'intégration partagé basé sur l'apprentissage contrastif supervisé et est spécifiquement conçu pour les scénarios d'intégration multimodale avec un chevauchement limité des caractéristiques.Il permet une prédiction précise du type cellulaire et une intégration transparente entre les différentes modalités et conditions expérimentales.Des tests effectués sur de multiples ensembles de données, notamment des PBMC sains, de la moelle osseuse, une thérapie CAR-T pour le lymphome et des tissus tumoraux, démontrent que le cadre surpasse les méthodes existantes tant en termes de qualité d'intégration que d'efficacité opérationnelle.
Titre de l'article :
CellFuse permet l'intégration multimodale des données de protéomique unicellulaire et spatiale
Lien vers l'article :
Parallèlement, les principales entreprises mondiales de biotechnologie et de santé accélèrent le déploiement de la technologie d'intégration de données multimodales unicellulaires, en se concentrant sur des scénarios clés tels que la translation clinique, le développement de médicaments et la médecine de précision, afin de transformer la recherche de pointe en applications concrètes. La société allemande BioNTech a appliqué cette technologie à l'immunothérapie des tumeurs et au développement de vaccins personnalisés. En intégrant le séquençage d'ARN unicellulaire, le profilage de l'expression protéique et les données de transcriptomique spatiale, elle analyse avec précision l'hétérogénéité cellulaire dans le microenvironnement tumoral, identifie les principaux sous-types de cellules immunitaires et les biomarqueurs associés, fournissant ainsi des données essentielles à la conception et à l'optimisation de vaccins antitumoraux personnalisés et améliorant significativement le ciblage et l'efficacité vaccinale.
Il est prévisible que, grâce aux progrès constants des technologies d'intégration multimodale, le décodage de la vie au niveau unicellulaire passera un jour de la vision à la réalité, insufflant un élan plus fort à l'avenir de la médecine de précision.