Command Palette
Search for a command to run...
Raffinement Quantique Piloté Par l'IA : l'université Carnegie Mellon Et d'autres Proposent AQuaRef, Le Premier À Utiliser Des Contraintes De Mécanique Quantique Pour Affiner Le Modèle Atomique Complet d'une protéine.
Pour comprendre les mécanismes moléculaires des processus vitaux, il nous faut d'abord observer la structure tridimensionnelle des macromolécules biologiques.La détermination des structures à l'échelle atomique est une tâche fondamentale de la biologie structurale et un socle important pour comprendre la fonction des protéines, révéler les mécanismes de régulation génétique et développer des médicaments ciblés.Qu’il s’agisse de réactions catalysées par des protéines, de transmission d’informations génétiques par les acides nucléiques ou de reconnaissance des antigènes par les anticorps, tous ces processus biologiques clés reposent sur des modèles structuraux précis pour être expliqués.
Actuellement, la cryo-microscopie électronique et la cristallographie aux rayons X sont les principales techniques expérimentales utilisées pour déterminer la structure des macromolécules biologiques, et une grande quantité de données structurales à haute résolution a été accumulée. Ces dernières années, les méthodes de prédiction computationnelle, telles qu'AlphaFold et RoseTTAFold, ont également connu des progrès significatifs, offrant des outils performants pour la modélisation structurale. Cependant, l'analyse expérimentale demeure indispensable à la découverte de structures inconnues et à la compréhension des interactions complexes.Dans le processus de résolution expérimentale de la structure, l'affinement du modèle atomique est une étape clé, proche de la phase finale. Son objectif est de construire un modèle moléculaire conforme aux lois de la stéréochimie et correspondant au mieux aux données expérimentales.Les logiciels d'affinage courants, tels que CCP4 et Phenix, s'appuient principalement sur des contraintes stéréochimiques dans des bases de données standard pour maintenir des longueurs et des angles de liaison raisonnables et réduire les conflits interatomiques.
Cependant, ces systèmes de contraintes présentent encore des limitations importantes. Ils ciblent principalement les structures covalentes et ne décrivent pas systématiquement les interactions non covalentes importantes telles que les liaisons hydrogène et l'empilement π. À basse résolution, cela peut conduire à des modèles s'écartant de l'état chimique réel. Lorsque de nouveaux ligands ou des liaisons uniques apparaissent dans la structure, la définition manuelle des paramètres est nécessaire pour l'affinement. De plus, des écarts géométriques raisonnables dus à l'environnement chimique local peuvent être interprétés à tort comme des anomalies par le système de contraintes et corrigés de force. Théoriquement,La mécanique quantique peut décrire les interactions intermoléculaires avec plus de précision, mais les macromolécules biologiques contiennent généralement des milliers, voire des dizaines de milliers d'atomes, ce qui rend l'informatique quantique complète extrêmement coûteuse.Par conséquent, la plupart des études existantes se limitent à des zones locales telles que les sites de liaison des ligands.
Pour remédier à ce problème, une équipe de recherche conjointe de l'Université Carnegie Mellon, de l'Université de Wrocław en Pologne et de l'Université de Floride, entre autres universités,Une méthode de raffinement quantique pilotée par l'IA, AQuaRef, est proposée.Cette méthode, basée sur l'apprentissage automatique AIMNet2 des fonctions de potentiel atomique, a été adaptée à la tâche d'affinement. Tout en offrant une efficacité de calcul comparable à celle des champs de force classiques, elle permet une meilleure approximation des résultats des calculs de mécanique quantique, ouvrant ainsi une nouvelle voie technique pour l'affinement quantique atomique des macromolécules biologiques.
Les résultats de cette recherche, intitulée « AQuaRef : raffinement quantique accéléré par l'apprentissage automatique des structures protéiques », ont été publiés dans Nature Communications.
Points saillants de la recherche :
* AQuaRef, basé sur la fonction potentielle d'apprentissage automatique AIMNet2, réalise pour la première fois un raffinement quantique d'un modèle atomique protéique complet.
* Lors de tests portant sur 61 modèles de microscopie à rayons X et cryo-électronique à basse résolution, AQuaRef a surpassé 57 modèles.
* Dans le cas de liaisons hydrogène courtes dans les protéines DJ-1 et YajL, AQuaRef peut déterminer les positions des protons en accord avec les données expérimentales sans intervention humaine.

Adresse du document :https://www.nature.com/articles/s41467-025-64313-1
Suivez notre compte WeChat officiel et répondez « AQuaRef » en arrière-plan pour obtenir le PDF complet.
Un ensemble de données d'un million d'échantillons pour l'entraînement de la fonction potentielle dans l'apprentissage automatique des peptides.
Cette étude vise à construire un modèle paramétré de la fonction potentielle d'un système peptidique en utilisant l'apprentissage automatique.Par conséquent, la conception de l'ensemble de données doit couvrir systématiquement trois dimensions : la composition chimique, l'espace conformationnel et les interactions intermoléculaires.
D'un point de vue chimique,Les chercheurs ont construit une petite base de données de peptides sous forme de chaînes SMILES, couvrant 20 acides aminés standard, 11 états de protonation, 3 modifications N-terminales et 4 modifications C-terminales.S’appuyant sur ces données, tous les monopeptides et dipeptides ont été énumérés, et un sous-ensemble de tripeptides et de tétrapeptides a été sélectionné aléatoirement. De plus, des peptides liés par des ponts disulfure et leurs analogues sélénisés ont été générés. Afin de couvrir l’ensemble de l’espace conformationnel, les chercheurs ont utilisé le logiciel OpenEye Omega pour un échantillonnage intensif des angles de torsion, sans imposer de restrictions sur les centres chiraux. Le modèle est ainsi applicable aux systèmes peptidiques de type D, de type L et aux systèmes stéréochimiques mixtes.
Des complexes composés de 2 à 4 peptides ont été construits, et leur orientation spatiale a été ajustée aléatoirement afin de simuler les interactions intermoléculaires. L'ensemble du processus de génération des données n'a fait appel à aucune séquence naturelle ni structure expérimentale afin d'éviter toute fuite de données. Pour limiter la complexité des calculs, le nombre total d'atomes (hydrogène compris) dans tous les peptides et leurs complexes a été fixé à 120.
Après avoir obtenu la conformation initialeLes chercheurs ont d'abord utilisé les champs de force GFN-FF pour mener des simulations de dynamique moléculaire afin d'échantillonner des structures hors d'équilibre.Il maintient la configuration globale proche de l'entrée initiale en la contraignant par des coordonnées cartésiennes, tout en libérant l'angle de torsion et les degrés de liberté intermoléculaires.
Par la suite, une stratégie d'apprentissage actif par interrogation collective a été mise en place : dans un premier temps, 500 000 échantillons initiaux ont été sélectionnés aléatoirement pour entraîner un système d'ensemble composé de quatre modèles. Quatre itérations ont ensuite été réalisées. À chaque itération, les échantillons ont été sélectionnés en fonction de l'incertitude des prédictions des modèles concernant l'énergie et les forces atomiques, et ces structures à forte incertitude ont été ajoutées à l'ensemble d'entraînement après calcul DFT. La dernière itération a introduit une optimisation guidée par l'incertitude, en privilégiant les structures limites présentant une forte incertitude de prédiction mais une faible énergie. Grâce à ce processus, un ensemble d'entraînement d'environ un million d'échantillons a finalement été obtenu, avec une moyenne d'environ 42 atomes.
Outre les données générées théoriquement, les chercheurs ont également examiné des structures expérimentales issues des bases de données RCSB et EMDB afin de valider le modèle. Les critères de sélection étaient les suivants : modèles à conformation unique ne contenant que des protéines, 1 000 à 10 000 atomes autres que l’hydrogène, résolution de 2,5 à 4 Å, score de conflit MolProbity inférieur à 50 et écarts de longueur et d’angle de liaison n’excédant pas quatre fois les valeurs standard.
AQuaRef : Approches de raffinement quantique pilotées par l’IA pour les systèmes macromoléculaires
AQuaRef commence par vérifier l'intégrité du modèle atomique d'entrée. Pour les atomes manquants dans la structure, le programme tente de les insérer automatiquement. Cependant, ce processus peut parfois introduire de nouveaux conflits d'encombrement stérique, notamment si le modèle original ne contient pas d'atomes d'hydrogène. Si les atomes manquants sont des éléments critiques, tels que des atomes de la chaîne principale, le modèle ne peut pas poursuivre l'affinement quantique. Si des conflits stériques importants ou des anomalies géométriques majeures sont détectés, une régularisation géométrique rapide, utilisant des contraintes stéréochimiques standard, est d'abord effectuée afin d'éliminer le problème avec des ajustements minimaux des positions atomiques.
Pour les données cristallographiques, l'affinement doit également tenir compte de la symétrie de la maille élémentaire et des interactions périodiques.Plus précisément, le programme développe le modèle en une supercellule en fonction de l'opérateur de symétrie du groupe spatial, puis la tronque en ne conservant que les copies symétriques dont la distance à l'atome de la copie principale se situe dans une plage définie. Ce processus est généralement inutile pour les structures observées en cryo-microscopie électronique.
Après l'ajout des atomes et l'expansion du modèle, le système entre dans le processus d'affinement standard du logiciel Q|R. L'architecture de base d'AQuaRef est fondamentalement identique à celle du modèle AIMNet2 de base, mais plusieurs ajustements importants ont été apportés pour l'affinement structural.
Premièrement, le modèle ne calcule pas explicitement les interactions coulombiennes et dispersives à longue portée, mais est directement entraîné pour reproduire l'énergie totale DFT-D4.En effet, dans le cadre du modèle de solvant implicite CPCM, l'interaction coulombienne est difficile à estimer précisément en utilisant uniquement la charge atomique, et l'interaction à longue portée est fortement atténuée par le milieu continu polarisable. De plus, les termes de dispersion à longue portée, dont le rayon de coupure est supérieur à 5 Å, contribuent très peu aux forces atomiques principales lors de l'affinement ; ils peuvent donc être négligés sans incidence sur la précision.
Deuxièmement, le modèle introduit un terme de répulsion exponentielle à courte portée explicite issu de GFN1-XTB, ce qui permet une meilleure stabilité lors du traitement de structures présentant des conflits d'encombrement stérique spatial.L'entraînement du modèle a été réalisé à partir de l'énergie, de la force atomique et de la charge atomique partielle de Hershfield calculées par la méthode B97M-D4/def2-QZVP. L'entraînement a débuté avec une initialisation aléatoire des poids, une taille de lot de 256 et un total de 1,5 million d'itérations. Tous les autres hyperparamètres ont été conservés par rapport à la configuration originale d'AIMNet2.
En ce qui concerne l'efficacité de calcul, comme le montre la figure ci-dessous...Dans le cadre AIMNet2, le temps de calcul de l'énergie et des forces atomiques, ainsi que l'utilisation maximale de la mémoire GPU, augmentent tous linéairement (O(N)) avec le nombre d'atomes dans le système.Pour un système protéique contenant environ 100 000 atomes, les calculs d’énergie et de force en un seul point ne prennent qu’environ 0,5 seconde ; sur un seul GPU NVIDIA H100 équipé de 80 Go de mémoire vidéo, des modèles contenant jusqu’à environ 180 000 atomes peuvent être traités.
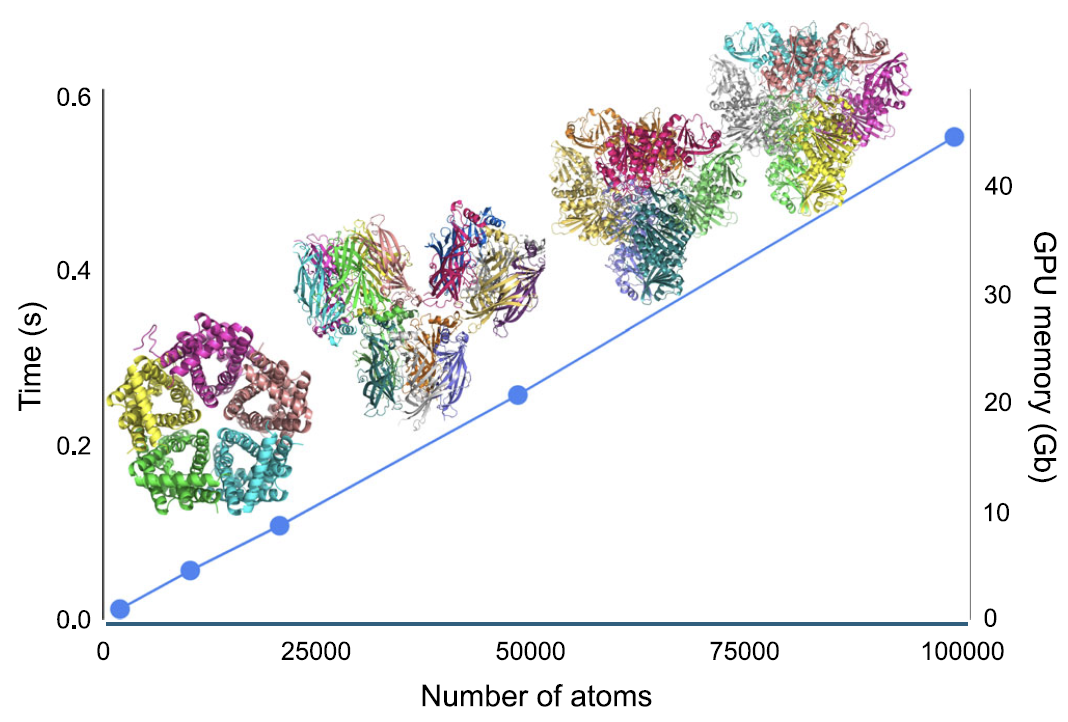
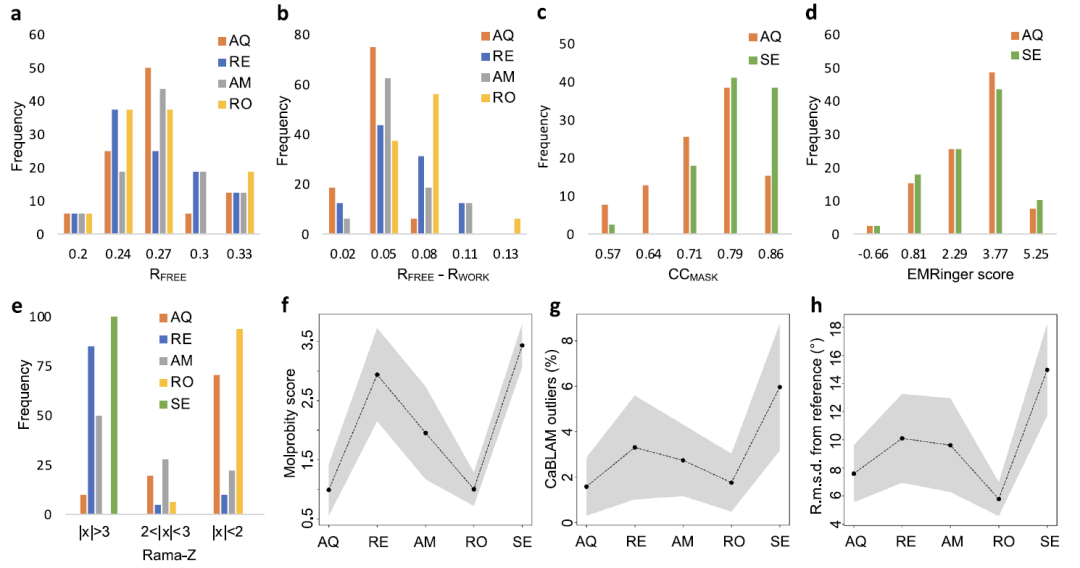
Validée par 41 analyses de cryo-microscopie électronique et 20 analyses de modèles aux rayons X, la structure locale d'AQuaRef a été optimisée à 2 Å.
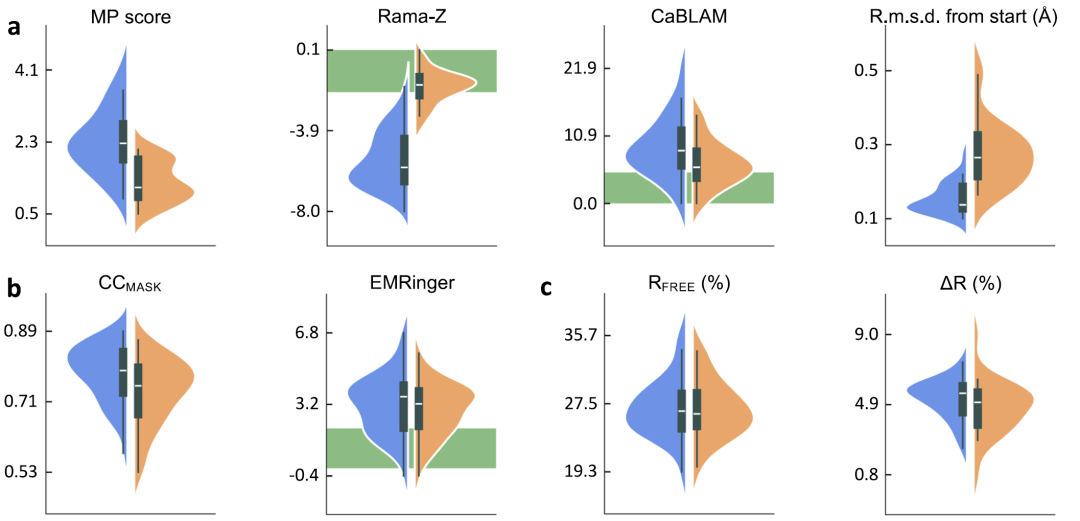
Pour évaluer les performances d'AQuaRef,Les chercheurs ont construit un ensemble de test comprenant 41 modèles de cryo-microscopie électronique, 20 modèles à basse résolution et 10 modèles à rayons X à ultra-haute résolution.Les 61 modèles basse résolution sont tous associés à des structures de référence homologues haute résolution. Lors de l'affinement, trois types de contraintes ont été définis à des fins de comparaison : les contraintes quantiques AIMNet2 (c.-à-d. AQuaRef), les contraintes géométriques standard et des contraintes supplémentaires, telles que les liaisons hydrogène et les structures secondaires, venant s'ajouter aux contraintes standard.
Les résultats sont présentés dans la figure ci-dessous.Le modèle à basse résolution raffiné quantiquement surpasse significativement les méthodes de contrainte traditionnelles dans les métriques géométriques telles que le score MolProbity et le score Z du diagramme de Ramachandran.Parallèlement, l'adéquation du modèle aux données expérimentales est restée globalement stable. Pour les structures obtenues par diffraction des rayons X, le surapprentissage a été légèrement réduit (la différence entre R<sub>work</sub> et R<sub>free</sub> était plus faible) ; pour les structures obtenues par cryo-microscopie électronique, le score CCmask a légèrement diminué tandis que le score EMRinger est resté pratiquement inchangé. Conjugués à l'amélioration globale de la qualité géométrique, ces résultats suggèrent une réduction du surapprentissage du modèle.
Bien que l'ajout de contraintes géométriques supplémentaires aux contraintes standard puisse améliorer la qualité du modèle, AQuaRef produit des géométries plus réalistes et se rapproche davantage du modèle de référence haute résolution. Dans certains cas, l'écart local entre les contraintes standard et la structure affinée quantiquement peut atteindre 2 Å.
L'étude a également comparé AQuaRef à plusieurs méthodes de raffinage courantes. Les résultats sont présentés dans la figure ci-dessous. AMBER, Rosetta et REFMAC5 ont été sélectionnés pour les données de diffraction des rayons X, tandis que Servalcat a été utilisé pour les données de cryo-microscopie électronique. Globalement,AQuaRef offre des performances Rfree légèrement supérieures et le plus faible degré de surapprentissage.Comparativement à Servalcat, les deux ont des scores EMRinger comparables, mais Servalcat a un score CCmask légèrement supérieur.
En termes de qualité géométrique,AQuaRef offre des performances similaires à Rosetta, mais surpasse nettement REFMAC5 et Servalcat.Rosetta présente une adéquation globale légèrement supérieure au modèle de référence, ce qui pourrait être lié au rayon de convergence plus important résultant de sa stratégie d'optimisation sans gradient. De plus, AQuaRef et Rosetta parviennent à générer des géométries de liaisons hydrogène raisonnables, suivis par AMBER, tandis que REFMAC5 et Servalcat sont pratiquement incapables de restituer ces détails avec précision.
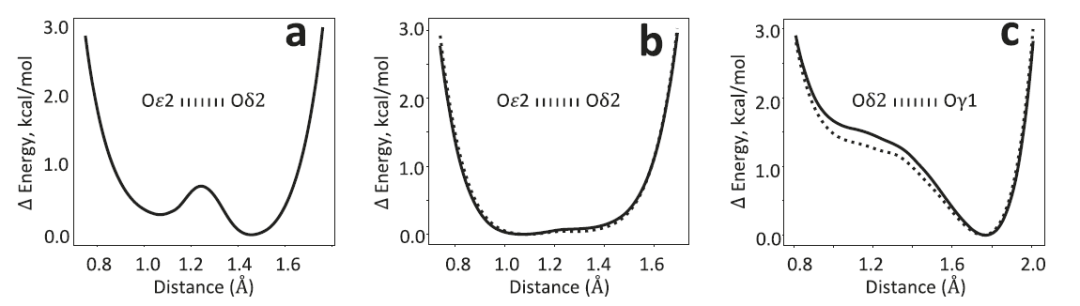
Lors de tests sur des systèmes de liaisons hydrogène courtes, les chercheurs ont utilisé la protéine DJ-1, associée à la maladie de Parkinson, et son homologue YajL comme exemples afin d'examiner la capacité d'AQaRef à gérer les états protonés. Les méthodes d'affinement classiques, contraintes par la stéréochimie des bases de données, entraînent souvent des écarts entre les longueurs de liaison et leurs valeurs réelles.Lorsque l'on utilise la structure diprotonée symétrique comme modèle initial pour l'affinement AQuaRef, les positions des protons et la géométrie des liaisons résultantes sont cohérentes avec les résultats de l'affinement non contraint.L'ajout de contraintes conventionnelles tend à rapprocher les longueurs de liaison des valeurs standard déprotonées de la base de données. Lorsque les données expérimentales sont tronquées à une résolution de 2 Å, réduisant considérablement les détails atomiques, AQuaRef parvient à reconstituer une structure quasi identique aux données originales à 1,15 Å, tandis que l'affinement par contraintes conventionnelles s'éloigne davantage de la configuration réelle. AQuaRef positionne le proton sur l'atome Oδ2 du résidu D24 de DJ-1, un résultat confirmé par les calculs d'énergie et les cartes de densité électronique différentielle.
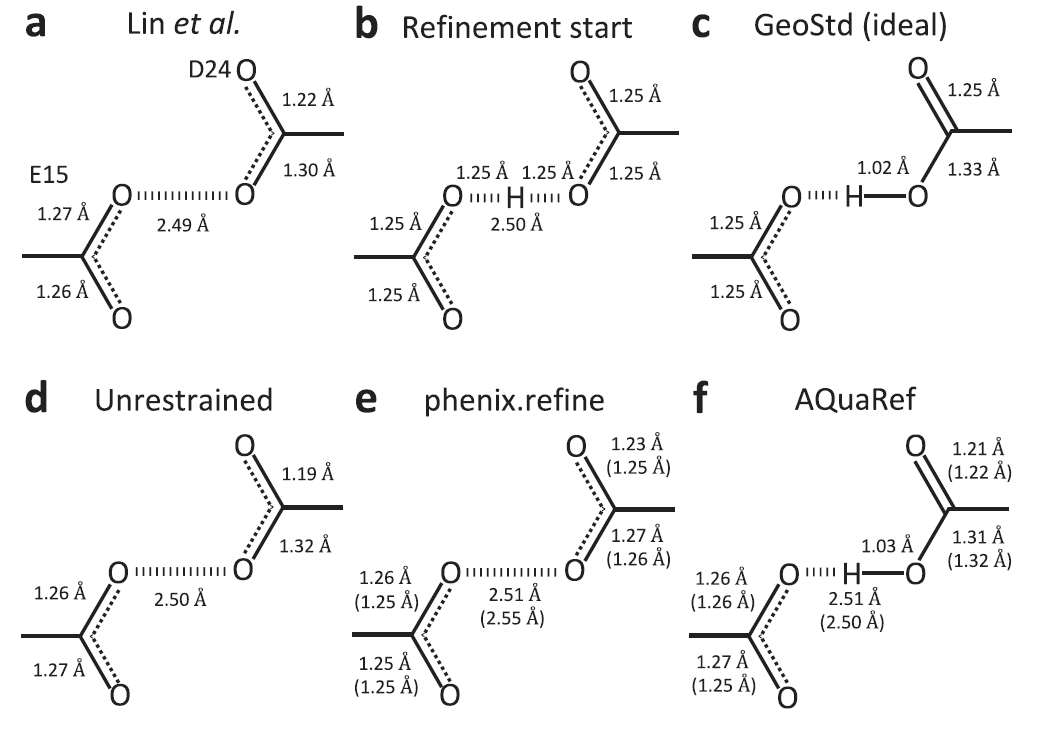
Dans la protéine YajL, les résultats d'affinement AQuaRef pour les deux liaisons hydrogène courtes E14/D23 sont cohérents avec l'affinement non contraint, indiquant que le proton est partagé par D23 et E14, ce qui correspond aux caractéristiques typiques d'une liaison hydrogène à faible barrière énergétique. Ceci diffère du cas de DJ-1, où le proton est principalement localisé sur un seul atome d'oxygène. La distribution d'énergie fournie par AIMNet2 montre une surface d'énergie potentielle relativement plate, ce qui signifie que la position du proton peut être ajustée librement dans les limites des données expérimentales. Parallèlement, la carte de densité électronique différentielle présente des pics nettement supérieurs à 3σ au voisinage de l'atome d'hydrogène, confortant ainsi cette interprétation structurale.
Avancées majeures dans la collaboration entre l'industrie, le monde universitaire et la recherche dans le domaine du raffinement quantique des protéines
Dans les domaines de pointe du raffinement quantique des protéines, de la construction de fonctions potentielles d'apprentissage automatique et de l'optimisation de modèles atomiques, de nombreuses équipes de recherche ont exploré sans relâche cette voie et ont réalisé une série de progrès. Par exemple,La méthode de réseau neuronal nn-tm fcc développée par l'équipe de l'Université d'Oxford peut construire des modèles de surface d'énergie potentielle de haute précision de fragments de résidus avec une précision quasi-quantique mécanique complète.Les erreurs quadratiques moyennes des calculs d'énergie et de forces atomiques sont respectivement inférieures à 1,0 kcal/mol et 1,3 kcal/(mol·Å). Grâce à cette méthode, les calculs d'énergie et de forces atomiques de 15 protéines représentatives peuvent être effectués en seulement 10 à 100 secondes, soit des milliers de fois plus rapidement que les calculs de mécanique quantique classiques.
Titre de l'article : Amélioration de la prédiction de la structure des protéines grâce aux potentiels issus de l'apprentissage profond
Lien vers l'article :https://www.nature.com/articles/s41586-019-1923-7
Une autre équipe de collaboration allemande a proposé l'algorithme quantique BF-DCQO, qui combine une stratégie itérative non variable avec le système de calcul quantique à piège à ions IonQ.Le temps de calcul pour un problème de repliement 3D impliquant 12 acides aminés a été réduit de 72 heures avec un cluster GPU traditionnel à environ 4,3 minutes.L'augmentation de vitesse a également été multipliée par mille.
Titre de la thèse : Algorithme quantique contrediabatique numérisé par champ de biais pour l’optimisation binaire d’ordre supérieur
Lien vers l'article :https://www.nature.com/articles/s42005-025-02270-3
Globalement, la combinaison de méthodes de mécanique quantique, de fonctions potentielles d'apprentissage automatique et de données structurelles expérimentales offre une nouvelle approche technique pour affiner les structures des macromolécules biologiques et devrait jouer un rôle plus stable dans des scénarios tels que la modélisation structurelle à basse résolution, l'analyse du mode de liaison des ligands et la recherche sur les sites fonctionnels.