Command Palette
Search for a command to run...
Le MIT a Développé Le Modèle Pichia-CLM Pour Apprendre Le « Langage » De l'ADN De La Levure, Ce Qui Pourrait Potentiellement Tripler Le Rendement En Protéines exogènes.

Dans les domaines biopharmaceutique et biotechnologique industriel, l'expression efficace des protéines recombinantes demeure un facteur déterminant des coûts de production et de la faisabilité des procédés. Des anticorps monoclonaux et antigènes vaccinaux aux préparations enzymatiques industrielles, même une légère augmentation des niveaux d'expression peut générer une valeur économique considérable.
Dans de nombreux systèmes d'expression,Pichia pastoris (Komagataella phaffii) est appréciée pour sa capacité de fermentation à haute densité, son système d'expression sécrétoire mature et ses excellentes capacités de traitement des protéines.Elle est devenue l'un des hôtes importants de la production industrielle. Cependant, un problème persistant qui affecte l'industrie est que, même si les séquences d'acides aminés sont parfaitement identiques, une simple modification des codons synonymes dans l'ADN codant peut entraîner des différences considérables dans les niveaux d'expression.
Ce phénomène est dû au biais d'utilisation des codons (BUC) : chez de nombreux organismes, certains codons synonymes sont utilisés de manière préférentielle. La sélection des codons synonymes influence le rendement protéique en agissant sur la transcription, la stabilité de l'ARNm, la traduction, le repliement des protéines, les modifications post-traductionnelles (MPT) et la solubilité.Par conséquent, « l’optimisation des codons » est devenue une étape clé dans l’expression des protéines exogènes.
Actuellement, divers outils et méthodes d'optimisation des codons basés sur les CUB de l'hôte ont été développés dans l'industrie, mais ces méthodes ne produisent pas toujours des constructions hautement expressives. Ces dernières années, avec le développement de l'intelligence artificielle, notamment des techniques de modélisation de séquences,Les chercheurs ont commencé à considérer les séquences génétiques comme une sorte de « langage » et tentent d'en apprendre les règles implicites à l'aide de méthodes similaires au traitement automatique du langage naturel.
Dans ce contexte,Une équipe de recherche du MIT a proposé un modèle de langage basé sur l'apprentissage profond, Pichia-CLM, pour l'optimisation des codons chez l'hôte industriel Pichia pastoris afin d'améliorer le rendement des protéines recombinantes.Contrairement aux méthodes traditionnelles qui s'appuient sur les métriques CUB (qui fournissent généralement un score global sans tenir compte du contexte de la séquence), Pichia-CLM exploite les données du génome hôte pour apprendre la correspondance entre acides aminés et codons de manière non biaisée. Des chercheurs ont validé expérimentalement Pichia-CLM sur six classes de protéines de complexité variable et ont systématiquement observé des rendements d'expression supérieurs à ceux obtenus avec quatre outils commerciaux d'optimisation des codons.
Les résultats de recherche connexes, intitulés « Pichia-CLM : un pipeline d'optimisation des codons basé sur un modèle de langage pour Komagataella phaffii », ont été publiés dans PNAS.
Points saillants de la recherche :
* Pichia-CLM utilise les données du génome hôte pour apprendre la correspondance entre les acides aminés et les codons de manière impartiale, en tenant compte non seulement des préférences de l'hôte, mais aussi de la dépendance à la position et des relations contextuelles à longue portée.
* Pichia-CLM a été validé expérimentalement sur six protéines de complexité variable, montrant systématiquement des rendements d'expression plus élevés.
* Les représentations vectorielles des acides aminés et des codons apprises par le modèle peuvent être regroupées en fonction de leurs propriétés physico-chimiques, ce qui indique que le modèle de langage peut capturer des schémas physiquement significatifs.
Adresse du document :
https://www.pnas.org/doi/10.1073/pnas.2522052123
Suivez notre compte WeChat officiel et répondez « Pichia pastoris » en arrière-plan pour obtenir le PDF complet.
Constituer un ensemble de données de séquences à grande échelle centré sur Pichia pastoris
Contrairement aux méthodes traditionnelles qui reposent sur des règles empiriques, l'idée centrale de Pichia-CLM est d'apprendre les modèles de codage directement à partir du génome hôte. À cette fin,L'équipe de recherche a constitué un ensemble de données de séquences à grande échelle centré sur Pichia pastoris.
Pour entraîner Pichia-CLM, les chercheurs ont collecté des données sur les acides aminés et les séquences codantes de deux variants de Pichia pastoris auprès de NCBI : CBS7435 et GS115. Ces données ont été complétées par des données précédemment réalisées dans leur laboratoire, notamment le séquençage et l’annotation du génome de GS115, K. phaffii (NRRL Y11430) et K. pastoris.Au total, environ 27 000 paires de données de séquences codant pour des acides aminés ont été utilisées.
Lors du traitement des données, les chercheurs ont tokenisé les acides aminés et les codons, et introduit des amorces (…). ),terminaison( ) et remplir ( L'ensemble de données est étiqueté afin de permettre au modèle de traiter des séquences de longueurs variables et de prendre en charge l'apprentissage par lots. De plus, il est divisé en ensembles d'entraînement et de test, environ 201 séquences TP3T étant utilisées pour évaluer la capacité prédictive du modèle sur des données non vues lors de l'entraînement.
Il convient de noter que cette méthode de construction des données n'introduit aucun « objectif d'optimisation » artificiel, mais repose entièrement sur des données génomiques naturelles. Ainsi, le modèle apprend les véritables préférences d'expression de l'hôte, plutôt que de définir artificiellement des règles approximatives, ce qui jette les bases d'améliorations de performance ultérieures.
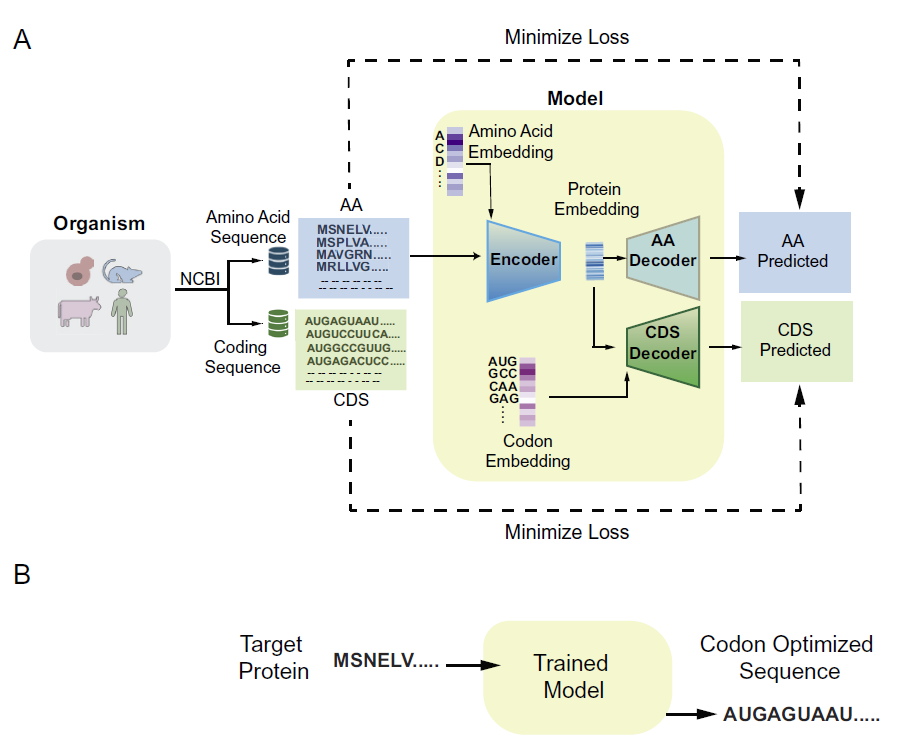
Pichia-CLM utilise une architecture encodeur-décodeur basée sur GRU.
Architecture du modèle
Le Pichia-CLM utilise une architecture encodeur-décodeur basée sur des unités récurrentes à porte (GRU).GRU est une architecture de réseau neuronal récurrent améliorée, conçue pour capturer les dépendances à court et à long terme dans les données séquentielles. En régulant le flux d'informations grâce à des mécanismes de contrôle, GRU atténue efficacement le problème de disparition du gradient, fréquent dans les RNN traditionnels. De plus, les performances de GRU sont comparables à celles des réseaux LSTM (Long Short-Term Memory), mais elle nécessite moins de paramètres et consomme moins de ressources de calcul, offrant ainsi des gains d'efficacité considérables pour de nombreuses tâches de modélisation de séquences.
Comparée à une autre architecture courante, Transformer, GRU offre une efficacité de calcul supérieure et une consommation de ressources moindre sur les ensembles de données de petite à moyenne taille.Des études ont montré qu'avec une taille de données d'environ 27 000 séquences, l'introduction d'un Transformer peut en fait augmenter inutilement la complexité, tandis que GRU permet d'obtenir un meilleur équilibre entre performance et efficacité.
Le modèle prend en entrée la séquence d'acides aminés d'une protéine et génère la séquence d'ADN correspondante à partir de motifs appris de la séquence d'acides aminés et de la séquence codante de l'hôte. L'architecture générale est illustrée dans la figure ci-dessous :
processus d'entraînement du modèle
Durant l'entraînement, les chercheurs ont utilisé un ensemble de validation (20% de l'ensemble d'entraînement) pour un arrêt précoce afin d'optimiser les paramètres. Simultanément, une sélection des hyperparamètres a été effectuée dans le but de minimiser la perte sur l'ensemble de validation (entropie croisée de classification parcimonieuse).L'optimisation des hyperparamètres utilise une stratégie d'optimisation globale appelée optimisation bayésienne, combinée à un code implémenté en interne par les chercheurs.
Plus précisément, le modèle fait intervenir les hyperparamètres suivants :
* Dimension d'incorporation des acides aminés
* Dimension d'intégration du codon
* Nombre d'unités dans la couche d'encodeur
* Taille de la couche entièrement connectée du codon dans le décodeur
* Taille de la couche entièrement connectée d'acides aminés dans le décodeur
Lors de l'entraînement du modèle, l'entrée du décodeur est la séquence encodée réelle (c'est-à-dire les codons réels). Lors de la phase de prédiction, le modèle utilise le codon prédit à la position précédente comme entrée pour la position suivante, réalisant ainsi une prédiction entièrement autorégressive. La prédiction de la séquence s'arrête lorsqu'un codon stop est rencontré.
Après avoir finalisé la sélection de l'architecture et validé la capacité prédictive du modèle sur l'ensemble de test, les chercheurs l'ont réentraîné sur l'ensemble des données complètes, en continuant d'appliquer une stratégie d'arrêt précoce pour éviter le surapprentissage. Ce modèle final a ensuite servi à concevoir les séquences codantes de protéines exogènes.
Pichia-CLM peut générer des constructions à forte production de protéines.
Dans la section consacrée à la validation expérimentale, l'équipe de recherche a sélectionné six protéines présentant différents niveaux de complexité pour les tests, notamment :
hormone de croissance humaine (hGH)
* Facteur de stimulation des colonies de granulocytes humains (hGCSF)
* VHH Nanobody 3B2 (34)
* Variante de sous-unité RBD du SARS-CoV-2 (RBD) modifiée par génie génétique (35)
* Albumine sérique humaine (HSA)
* Anticorps monoclonal IgG1 trastuzumab (Trast)
Performance de Pichia-CLM dans l'amélioration de la sécrétion protéique chez Pichia pastoris
d'abord,Les chercheurs ont sélectionné trois protéines d'origine humaine de tailles et de complexités variables : hGH, hGCSF et HSA, et ont comparé les différences de rendement de sécrétion protéique (titre) entre les constructions génétiques générées à l'aide de Pichia-CLM et leurs séquences codantes natives.Globalement, pour des protéines telles que hGH et hGCSF, l'augmentation du rendement était d'environ 25% ; tandis que pour HSA, une augmentation significative d'environ 3 fois a été observée.
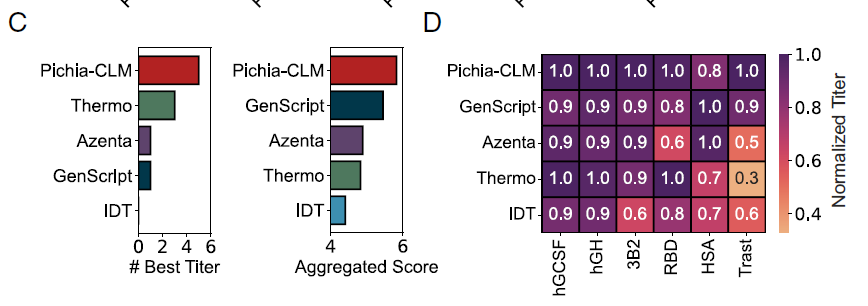
Par la suite, les chercheurs ont comparé Pichia-CLM à quatre outils commerciaux d'optimisation des codons : Azenta, IDT, GenScript et Thermo Fisher (Thermo), en évaluant six protéines à l'aide de deux métriques :
* MeilleurTitre : Le nombre de protéines ayant le titre le plus élevé obtenu par une méthode particulière.
* Score agrégé : La somme des titres relatifs des différentes protéines (normalisée par rapport à la valeur maximale).
Dans l'ensemble,Pichia-CLM a surpassé les algorithmes commerciaux dans les deux métriques (figure C ci-dessous) ; il a atteint le titre le plus élevé dans 5 des 6 protéines, avec seulement une légère diminution du score global (environ 0,2) dans HSA en raison d'un titre légèrement inférieur (figure D ci-dessous).
Évaluation des caractéristiques de la séquence génétique
Après avoir validé les performances de Pichia-CLM dans la production de protéines exogènes, les chercheurs ont analysé plus en détail les caractéristiques de séquence génétique de différentes constructions conçues.À l'instar d'autres modèles de langage protéique déjà publiés, l'optimisation des codons repose généralement sur une ou plusieurs métriques de biais d'utilisation des codons (BUC) pour sa conception ou son évaluation. Par conséquent, cette étude a utilisé les données de six protéines tests afin d'évaluer la corrélation entre ces métriques BUC et le rendement protéique.
Les résultats ont montré qu'aucun de ces indicateurs n'affichait une corrélation élevée et constante avec le rendement pour différentes protéines. Par exemple, dans le cas de l'HSA (comme illustré dans la figure A ci-dessous), la corrélation positive maximale avec la volatilité des codons et la distribution de fréquence des codons (CFD) n'était que de 0,43, tandis que la corrélation négative maximale avec le score de paires de codons (CPS) n'était que de 0,25.
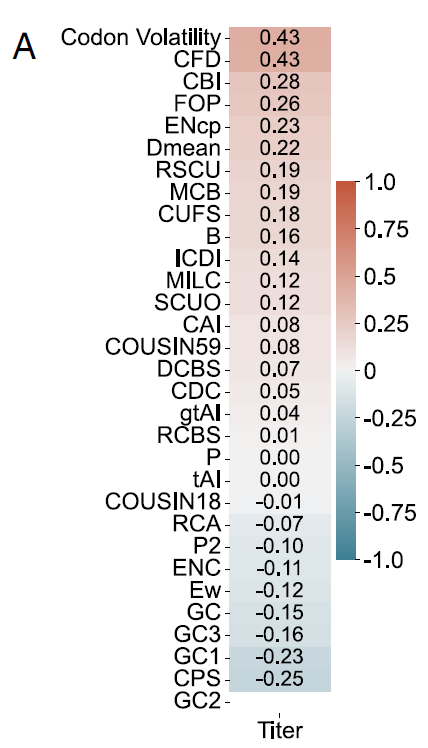
Les métriques CUB globales calculées sur la base de la séquence entière présentent des limitations importantes pour la caractérisation des caractéristiques liées à la production de protéines exogènes.Cela démontre une fois de plus la nécessité de nouvelles métriques d'évaluation pour évaluer les outils d'optimisation des codons, combinées à une validation expérimentale rigoureuse de diverses protéines – un résultat qui remet directement en question le fondement théorique de l'optimisation traditionnelle des codons.
Évaluation des caractéristiques de séquence
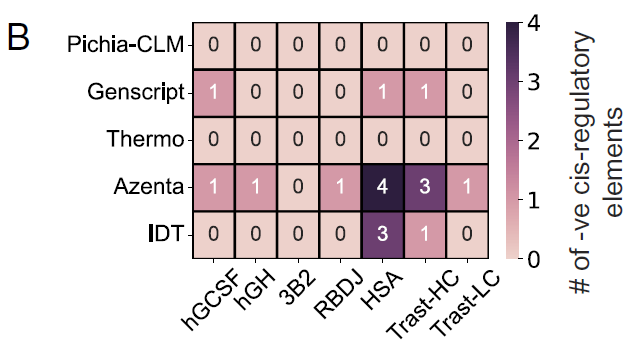
Les chercheurs ont également évalué la présence d'éléments cis-régulateurs négatifs dans différentes constructions optimisées pour les codons, qui pourraient interférer avec les mécanismes de régulation de l'hôte et devraient donc être évités autant que possible dans les séquences d'ADN exogènes.
Parmi les 6 protéines testées,Aucun élément cis-régulateur négatif n'a été détecté dans les constructions conçues à l'aide de Pichia-CLM ; en revanche, GenScript contenait un élément cis-régulateur négatif dans trois des six protéines ; Azenta et IDT ont généré des séquences contenant trois à quatre éléments de ce type dans au moins une protéine.Comme le montre la figure B :
Les chercheurs ont également analysé les performances de Pichia-CLM sur 52 protéines liées aux biotechnologies, et les résultats ont montré :La séquence protéique de 75% ne contient aucun élément cis-régulateur négatif, tandis que le 25% restant contient au maximum deux éléments de ce type.En revanche, l'algorithme commercial le plus performant, GenScript, a tout de même produit des constructions contenant de 3 à 6 éléments cis-régulateurs négatifs dans des protéines d'environ 15%, comme le montre la figure C ci-dessous :
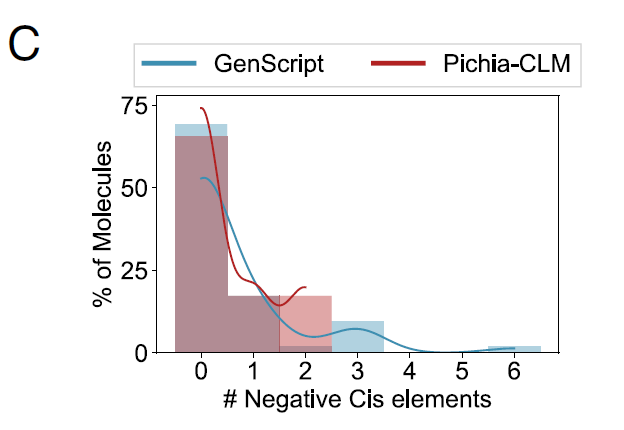
En résumé, ces résultats démontrent que Pichia-CLM peut non seulement générer des constructions protéiques à haut rendement, mais aussi apprendre des caractéristiques clés de la séquence génétique et atteindre un équilibre entre de multiples facteurs, concevant ainsi des séquences codantes robustes adaptées à l'expression par l'hôte.
L'IA accélère l'industrialisation de la production de protéines
Dans l'industrie biopharmaceutique, l'amélioration de l'efficacité de la production de protéines a toujours été un facteur déterminant du succès ou de l'échec de la recherche et du développement, ainsi que de leur commercialisation. Des anticorps monoclonaux aux vaccins recombinants, en passant par diverses protéines de fusion et préparations enzymatiques, la demande du marché ne cesse de croître, et les exigences en matière de rendement, de stabilité et de constance sont de plus en plus élevées.
Pour atteindre cet objectif, l'industrie a développé un système d'optimisation à plusieurs niveaux : au niveau de l'hôte, outre les souches traditionnelles d'E. coli et de Saccharomyces cerevisiae, Pichia pastoris et les cellules de mammifères sont devenues les principales plateformes de production en raison de leurs capacités supérieures de modification post-traductionnelle et de leur efficacité d'expression ; au niveau de la conception moléculaire, outre l'optimisation des codons, cela inclut la régulation de la force du promoteur, le criblage des peptides signaux, l'ingénierie structurale de l'ARNm et l'optimisation du repliement des protéines et de la voie de sécrétion ; et au niveau du procédé, la fermentation à haute densité, l'optimisation de la stratégie d'alimentation et le contrôle des paramètres du bioréacteur jouent également un rôle décisif dans le rendement final.
En dehors de ce système,Une nouvelle technologie de « décellularisation » émerge rapidement : la synthèse protéique acellulaire (CFPS).Cette technologie contourne le processus de croissance cellulaire et exploite directement le système de transcription et de traduction des lysats cellulaires pour obtenir une expression protéique rapide. Elle est largement utilisée dans le développement et la production d'anticorps, d'enzymes et même de conjugués anticorps-médicament. Cependant, le système CFPS lui-même est un système multivarié très complexe impliquant des dizaines de composants tels que des matrices d'ADN, des systèmes enzymatiques, des donneurs d'énergie, des acides aminés et des environnements ioniques. Son espace de combinaisons est extrêmement vaste et les méthodes d'optimisation classiques, basées sur l'expérience, peinent souvent à atteindre un équilibre optimal entre coût et rendement.
Dans ce contexte, l'optimisation automatisée pilotée par l'IA révèle un potentiel de rupture. Récemment, OpenAI, en collaboration avec Ginkgo Bioworks, entreprise leader en biologie synthétique, a publié des résultats de recherche novateurs.Le « système d'automatisation en boucle fermée » construit sur le modèle de langage étendu GPT-5 a réussi à réaliser une double optimisation de la technologie de synthèse protéique acellulaire (CFPS) : réduire le coût total de production de la technologie de 401 TP3T, réduire considérablement les coûts des réactifs de 571 TP3T et améliorer le titre de synthèse protéique de 271 TP3T.
À l'avenir, des approches similaires seront étendues à un plus large éventail de scénarios de bioproduction. De l'optimisation des voies métaboliques dans les usines cellulaires au contrôle en temps réel des processus de fermentation et à la conception intelligente de constructions d'expression, l'intelligence artificielle s'intègre progressivement à tous les aspects de la production de médicaments protéiques.
Références :
1.https://www.pnas.org/doi/10.1073/pnas.2522052123
2.https://phys.org/news/2026-02-ai-yeast-dna-language-boost.html#google_vignette
3.https://mp.weixin.qq.com/s/Qkl6j9HcFB7W_Y5Xh-9BCw