Command Palette
Search for a command to run...
À Partir De 14 000 Ensembles De Données Réelles, L’université De Washington/Microsoft Et D’autres Ont Proposé GigaTIME Pour Créer Un Atlas Panoramique Du Microenvironnement Immunitaire tumoral.
Dans le contexte évolutif du cancer, le microenvironnement immunitaire tumoral domine non seulement la croissance, l'invasion et la métastase des cellules cancéreuses, mais influence aussi profondément la réponse au traitement et le pronostic final du patient. Il ne s'agit pas d'une action isolée des cellules cancéreuses, mais d'un écosystème hautement dynamique : cellules immunitaires, fibroblastes, cellules endothéliales et autres cellules interagissent et s'intègrent conjointement à la matrice extracellulaire, dont la structure et la fonction ont été remodelées, formant ainsi un réseau pathologique précis et complexe.
La clé pour déchiffrer ce réseau réside dans la compréhension des états fonctionnels et des interactions des cellules, et les niveaux d'activation de protéines spécifiques constituent des « codes moléculaires » cruciaux. Traditionnellement,L'immunohistochimie (IHC) est devenue un outil classique pour déchiffrer les codes grâce à sa capacité à visualiser la localisation des protéines.Par exemple, le marquage PD-L1 est largement utilisé pour identifier le statut des points de contrôle immunitaire et prédire l'efficacité de l'immunothérapie. Cependant, l'IHC ne permet d'obtenir des informations que sur une seule protéine à la fois, ce qui rend difficile la reconstitution de l'écologie réelle de la coexistence de plusieurs protéines. Ceci constitue un obstacle majeur à une compréhension plus approfondie du mécanisme de dialogue entre les cellules tumorales et immunitaires.
Pour pallier cette limitation, la technologie d'immunofluorescence multiplex (mIF) a été développée. Elle permet de visualiser simultanément la distribution spatiale de plusieurs protéines sur une même coupe de tissu, préservant ainsi intégralement les informations contextuelles de la structure tissulaire.Cependant, cette technologie est coûteuse et son processus est complexe, la coloration, l'imagerie et l'analyse étant toutes extrêmement chronophages.Cela rend difficile l'accumulation de données à grande échelle et entrave la transposition clinique.
À l'inverse, les coupes colorées à l'hématoxyline-éosine sont largement disponibles et peu coûteuses en milieu clinique. Bien qu'elles ne révèlent pas directement l'activité protéique, elles préservent parfaitement la structure globale du tissu et les détails de la morphologie cellulaire. Les caractéristiques cachées qu'elles présentent peuvent refléter indirectement l'état fonctionnel des cellules, mais ces motifs subtils et complexes dépassent souvent les limites de la perception visuelle humaine.
Ces dernières années, les avancées en intelligence artificielle ont ouvert de nouvelles perspectives. Grâce à un pré-entraînement sur d'immenses quantités d'images pathologiques, l'IA a démontré de puissantes capacités d'analyse visuelle et d'extraction de caractéristiques. Ceci soulève une question essentielle : l'IA peut-elle être utilisée pour « décoder » les informations d'activation des protéines à partir d'images H&E facilement disponibles, informations dont l'acquisition nécessitait auparavant la coûteuse technologie mIF ?
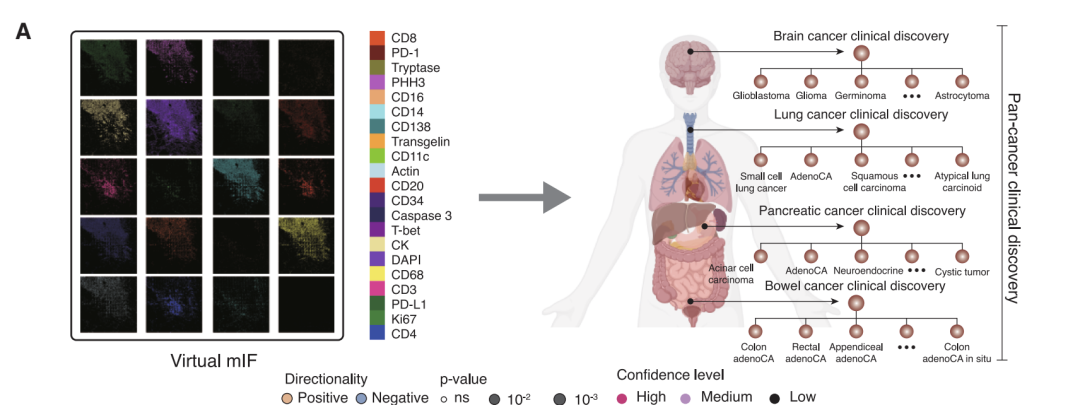
Partant de ce raisonnement,Une équipe de recherche composée de Microsoft Research, de l'Université de Washington et de Providence Genomics a proposé GigaTIME, un cadre d'intelligence artificielle multimodal.Grâce à une technologie d'apprentissage multimodal avancée, il est possible de générer des cartes mIF virtuelles à partir de coupes H&E conventionnelles. L'équipe de recherche l'a appliquée à une cohorte de plus de 14 000 patients atteints de cancer au Providence Medical Center aux États-Unis, couvrant 24 types de cancer et 306 sous-types, générant ainsi près de 300 000 images mIF virtuelles et permettant une modélisation systématique du microenvironnement immunitaire tumoral au sein d'une population vaste et diversifiée.
Les résultats de cette recherche, intitulée « L’IA multimodale génère une population virtuelle pour la modélisation du microenvironnement tumoral », ont été publiés dans la revue Cell.
Points saillants de la recherche :
* GigaTIME utilise une IA multimodale pour convertir les lames de pathologie H&E en données de protéomique spatiale, générant des populations virtuelles contenant des états cellulaires à partir de lames H&E de routine.
* Soutient la découverte clinique à grande échelle et la stratification des patients, et révèle de nouveaux modèles d'activation protéique spatiaux et combinatoires.

Adresse du document :https://www.cell.com/cell/fulltext/S0092-8674(25)01312-1
Suivez notre compte WeChat officiel et répondez « Vaccinations multiples » en arrière-plan pour obtenir le PDF complet.
Autres articles sur les frontières de l'IA :
https://hyper.ai/papers
Jeux de données : Construire une boucle complète, de l’entraînement à l’application
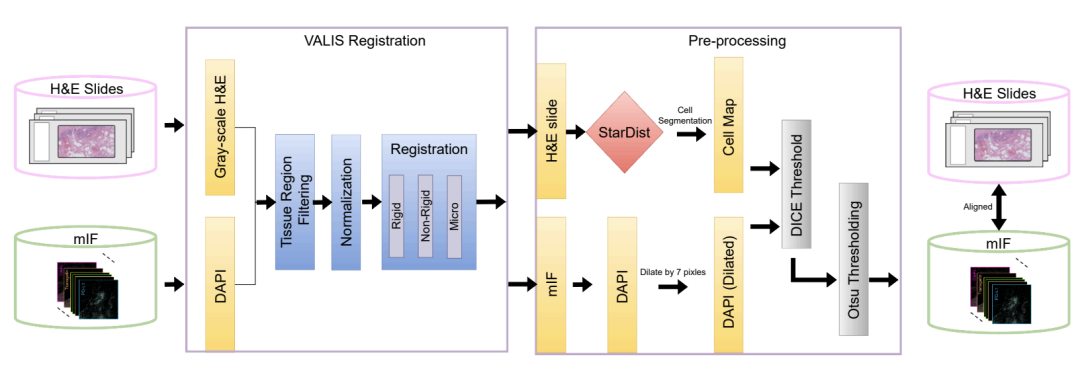
L'entraînement d'un modèle nécessite d'abord de résoudre une contradiction fondamentale : la coloration H&E, largement disponible et peu coûteuse en pratique clinique, ne permet pas de révéler directement l'activité des protéines, tandis que la technologie mIF, capable de révéler les relations spatiales entre plusieurs protéines, est onéreuse et complexe, ce qui rend son déploiement à grande échelle difficile. Pour construire un modèle d'IA qui relie ces deux techniques d'imagerie,L'équipe de recherche a utilisé la plateforme COMET pour collecter 441 images mIF à partir de 21 sections colorées H&E.Comme le montre la figure ci-dessous, ces images couvrent un total de 21 biomarqueurs clés, allant des protéines nucléaires telles que DAPI et PHH3, aux protéines de surface telles que CD4 et CD11c, en passant par les protéines cytoplasmiques telles que CD68. Elles fournissent des preuves importantes pour l'analyse de la composition, de l'état fonctionnel et de l'activité des cellules immunitaires dans le microenvironnement tumoral.

Après l'obtention des images appariées, le principal défi consiste à en extraire des données d'entraînement de haute qualité. À cette fin, comme illustré ci-dessous, l'équipe a conçu un flux de traitement rigoureux : tout d'abord, l'outil VALIS est utilisé pour aligner précisément l'image H&E et l'image mIF au niveau du pixel ; ensuite, l'algorithme StarDist est utilisé pour identifier et segmenter chaque cellule de l'image ; enfin, la région de l'image présentant la meilleure qualité d'alignement est sélectionnée en fonction du coefficient de Dice.
À travers plusieurs niveaux de contrôle de qualitéL'équipe a sélectionné 10 millions de cellules de haute qualité parmi les données initiales contenant 40 millions de cellules et les a divisées en ensemble d'entraînement, ensemble de validation et ensemble de test indépendant.De plus, l'étude a introduit des échantillons de cancer du sein et du cerveau provenant de microarrays tissulaires comme ensemble de validation externe. Ces échantillons différaient significativement des données d'entraînement par leur structure et leur morphologie tissulaires : ils se présentaient sous forme de petits blocs de tissu cylindriques séparés par des zones vides, contrairement aux grandes tranches de tissu continues des données d'entraînement. Ceci a permis de tester efficacement la capacité de généralisation du modèle face à de nouveaux types d'échantillons et à des types de cancers inconnus.
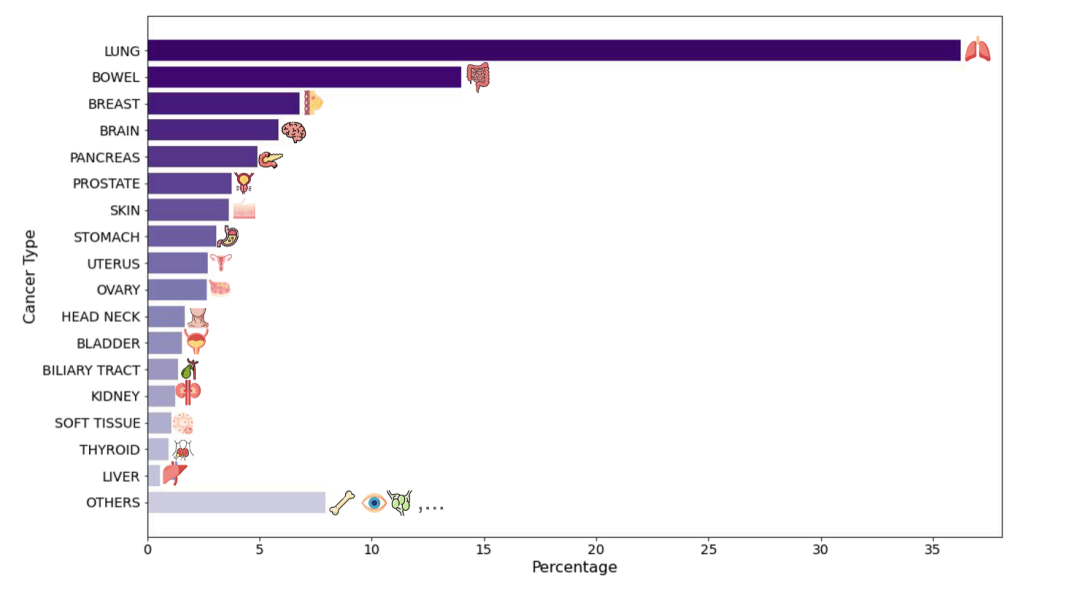
Au niveau de l'application du modèle, l'étude a construit deux cohortes de population virtuelles à grande échelle et complémentaires.La première cohorte provient du réseau clinique de Providence Health, un groupe de santé américain, et comprend des coupes histologiques (coloration H&E) de 14 256 patients atteints de cancer, répartis dans 51 hôpitaux et plus de 1 000 cliniques. Elle couvre 24 principaux types de cancer et 306 sous-types, et intègre de nombreuses informations cliniques telles que des biomarqueurs génomiques, le stade pathologique et le suivi de la survie. La valeur unique de cet ensemble de données réside dans son réalisme : une population de patients très diversifiée, couvrant tout le spectre des stades de la maladie, des stades précoces aux stades avancés, reflétant fidèlement la complexité de la pratique clinique.
La deuxième cohorte a été extraite de la base de données publique Cancer Genome Atlas (TCGA).L'étude a porté sur 10 200 lames histologiques colorées à l'hématoxyline-éosine, provenant principalement de cas chirurgicaux précoces non traités. Ces deux cohortes présentaient un contraste marqué quant à l'origine des patients, le stade de la maladie et le contexte clinique. Cette conception différenciée offrait des conditions optimales pour valider la fiabilité et la généralisabilité du modèle : la cohérence et la robustesse des conclusions biologiques obtenues à travers des ensembles de données aussi divers suggèrent fortement son large potentiel clinique.
GigaTIME : Construire des ponts intelligents alliant forme et fonction
Le modèle GigaTIME s'attaque directement à un obstacle majeur dans la recherche sur le microenvironnement immunitaire tumoral : la technologie mIF, coûteuse et à faible débit, est difficile à généraliser, tandis que les images de coloration H&E obtenues en routine clinique ne reflètent pas directement l'activité fonctionnelle des protéines. Ce modèle utilise l'intelligence artificielle pour apprendre et générer des images mIF virtuelles à partir d'images H&E, offrant ainsi une voie viable pour une analyse systématique et peu coûteuse du microenvironnement immunitaire tumoral à l'échelle de la population.
Le modèle utilise un cadre encodeur-décodeur patchwork soigneusement conçu, dont le noyau est construit sur un réseau imbriqué en forme de U.L'avantage de cette architecture réside dans sa capacité à capturer simultanément les subtilités locales et la structure organisationnelle globale d'une image. Plus précisément, l'encodeur extrait des représentations de caractéristiques multiniveaux à partir d'un patch d'image H&E de 256 × 256 pixels par convolution et sous-échantillonnage ; le décodeur reconstruit ensuite ces caractéristiques abstraites en une image mIF virtuelle avec une résolution spatiale par suréchantillonnage et fusion de caractéristiques. Cette conception permet au modèle de se concentrer à la fois sur la morphologie fine des cellules individuelles et sur les schémas d'organisation des populations cellulaires.
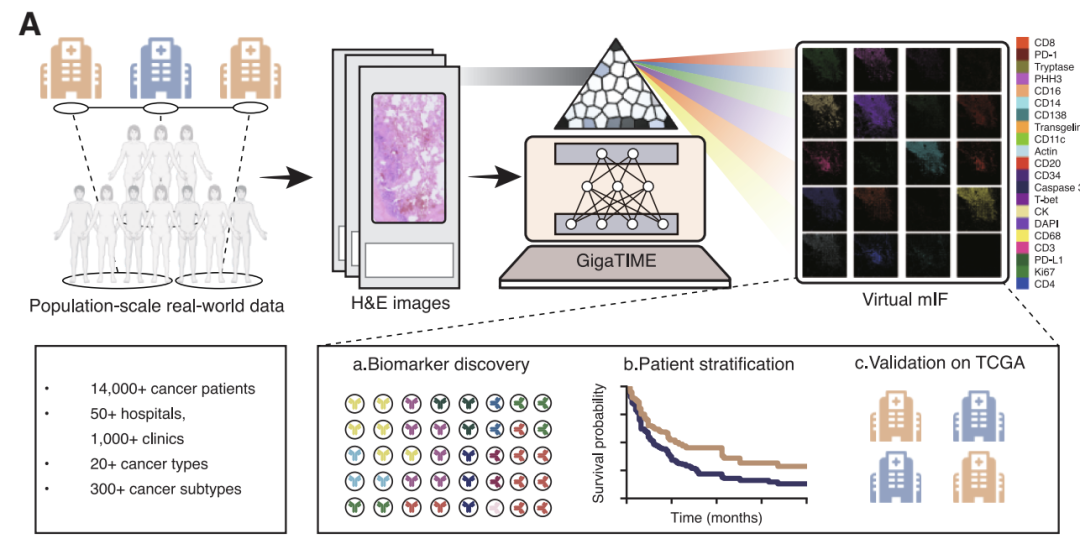
Au niveau des résultats, la conception du modèle témoigne d'une prise en compte approfondie des questions biologiques.Pour chacun des 21 canaux protéiques prédéfinis, GigaTIME effectue une prédiction de classification binaire pour chaque pixel de l'image d'entrée.Le système détermine si une protéine spécifique est activée à un emplacement donné, générant ainsi une carte d'activité protéique à l'échelle du pixel. Ces prédictions locales peuvent être assemblées de manière continue pour reconstruire une image mIF virtuelle de la totalité de la coupe tissulaire. Ceci permet le calcul de divers indicateurs quantitatifs, tels que la densité d'activation et le profil de distribution spatiale de protéines spécifiques dans la région tumorale, fournissant ainsi une base de données solide pour des analyses à haut débit ultérieures et des études de corrélation clinique.
Pour garantir un apprentissage efficace du modèle, la stratégie d'entraînement a été systématiquement optimisée.La fonction de perte combine astucieusement la perte de Dice et la perte d'entropie croisée binaire : la première vise à garantir la cohérence globale entre la région active prédite et la région réelle en termes de contour spatial, tandis que la seconde vise à améliorer la précision de la classification de chaque pixel. La synergie entre les deux assure à la fois une reconstruction précise du motif spatial global et une fiabilité au niveau des détails. Le modèle a été entraîné pendant 250 époques sur 8 GPU NVIDIA A100 avec une taille de lot de 16 et un taux d'apprentissage de 0,0001. Tous les hyperparamètres clés ont été déterminés par débogage système à partir des résultats de l'ensemble de validation.
Il est particulièrement important de souligner que le succès du modèle dépend fortement de la qualité des données d'entraînement.L'équipe de recherche a utilisé des procédures rigoureuses d'enregistrement d'images, de segmentation cellulaire et de contrôle de la qualité.Dix millions de cellules de haute qualité ont été sélectionnées parmi les données initiales massives pour l'entraînement, garantissant ainsi que le modèle apprenne des correspondances intermodales robustes, fiables et biologiquement pertinentes, plutôt que des régularités statistiques superficielles ou des modèles bruités.
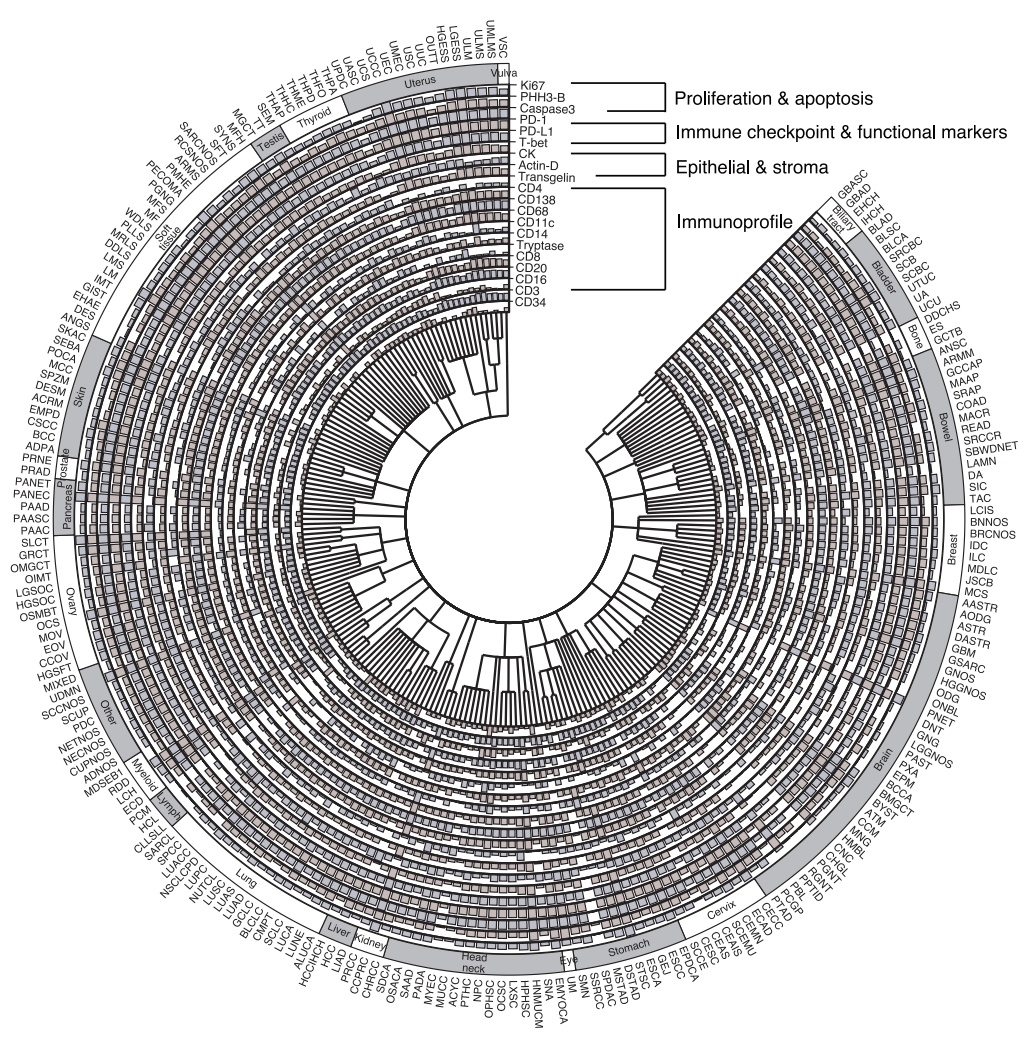
Résultats à grande échelle basés sur près de 300 000 images virtuelles : GigaTIME révèle 1 234 associations cliniques
Afin d'évaluer de manière exhaustive les performances et la valeur de GigaTIME, l'équipe de recherche a conçu un schéma d'évaluation systématique selon deux dimensions : la validation technologique et les résultats cliniques.
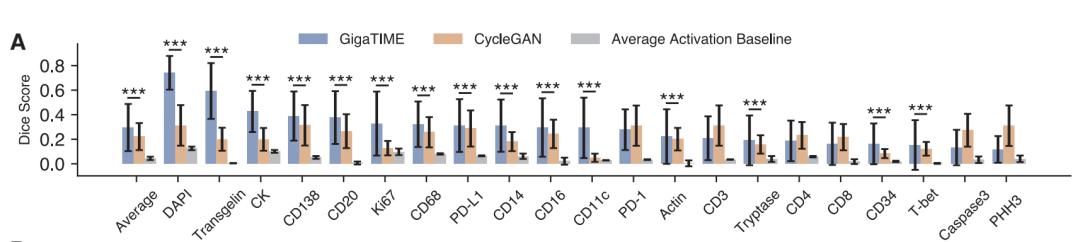
En termes de vérification technique,L'étude a évalué les capacités de conversion d'images du modèle à trois niveaux : pixel, cellule et tranche.Au niveau du pixel, GigaTIME surpasse nettement le modèle de référence CycleGAN sur 15 des 21 canaux protéiques. Par exemple, sur le canal DAPI, GigaTIME atteint un coefficient de Dice de 0,72, dépassant largement les 0,12 du modèle statistique de référence.
Au niveau cellulaire,GigaTIME a atteint une corrélation de 0,59 sur le canal DAPI, tandis que CycleGAN n'a atteint que 0,03, se rapprochant d'un niveau aléatoire.
Au niveau de la trancheLe coefficient de corrélation du canal DAPI de GigaTIME atteint 0,98, avec une moyenne de 0,56 sur l'ensemble des canaux, tandis que celui de CycleGAN est proche de 0. Ces résultats démontrent que l'apprentissage supervisé basé sur des données appariées de haute qualité est crucial pour une conversion intermodale précise.
En termes de résultats cliniques, l'étude a utilisé près de 300 000 images mIF virtuelles provenant de 14 256 patients.L'association entre l'expression de protéines virtuelles et 20 biomarqueurs cliniques a été analysée de manière systématique.Après des tests statistiques rigoureux et de multiples corrections, un total de 1 234 associations significatives ont été identifiées, réparties sur trois niveaux : pan-cancer, type de cancer et sous-type de cancer.
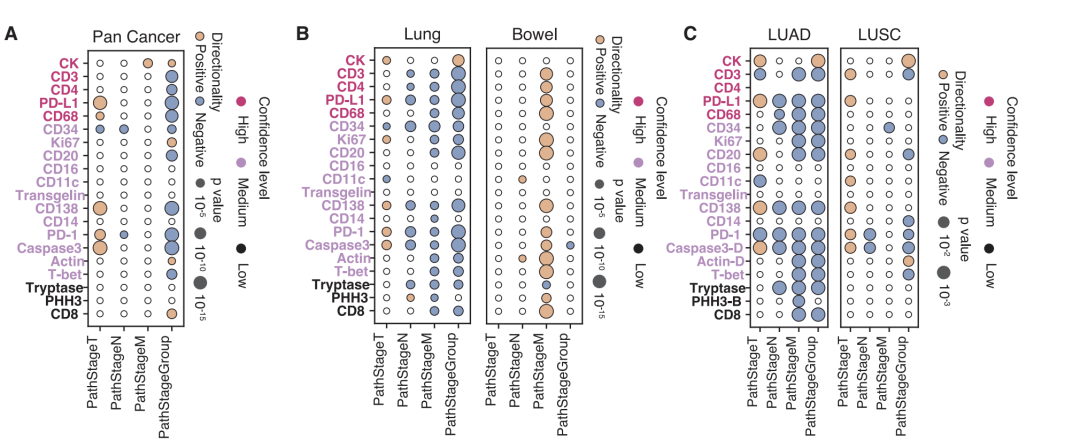
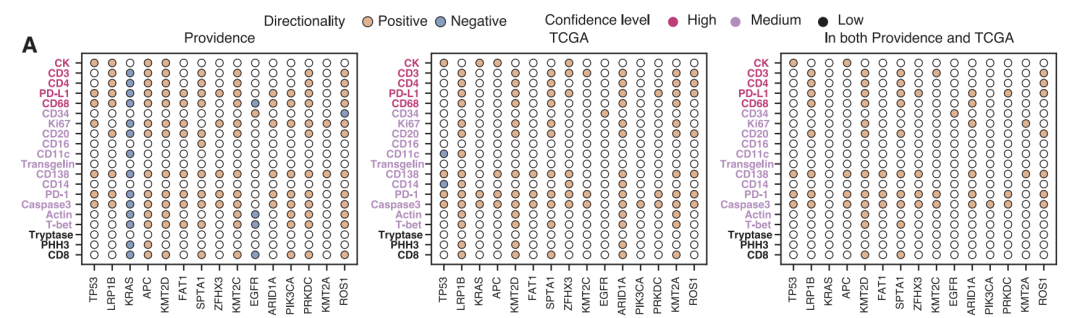
Parmi les 175 associations identifiées dans l'analyse pancancéreuse, une charge mutationnelle tumorale élevée et une forte instabilité microsatellitaire étaient significativement associées à une activation accrue de plusieurs marqueurs d'infiltration immunitaire (CD138, CD20, CD68 et CD4), ce qui est cohérent avec un mécanisme d'activation immunitaire induit par l'antigène. De nouveaux indices ont également été mis en évidence : les mutations de KMT2D présentaient une forte corrélation positive avec les marqueurs immunitaires, suggérant une potentielle promotion de l'infiltration immunitaire ; tandis que les mutations de KRAS présentaient une corrélation négative, reflétant un phénotype de rejet immunitaire. Dans certains types et sous-types de cancer, le modèle a révélé de nombreuses associations spécifiques. Par exemple, la forte corrélation entre les mutations de T-bet et de TP53 dans le cancer du cerveau n'a pas été détectée à l'échelle pancancéreuse, ce qui pourrait être lié au microenvironnement immunitaire particulier du système nerveux central. L'analyse des sous-types de cancer du poumon a montré que les mutations PRKDC dans l'adénocarcinome pulmonaire étaient plus fortement associées aux marqueurs de réponse immunitaire que dans le carcinome épidermoïde, confirmant l'importance d'interpréter les données en tenant compte du contexte histologique.
L'étude a également validé la valeur des données virtuelles dans les résultats cliniques.L'analyse a révélé une corrélation positive entre la taille de la tumeur primitive (stade T) et les points de contrôle immunitaire ainsi que les marqueurs d'invasion. Cependant, cette association s'inverse aux stades avancés, suggérant que les tumeurs avancées pourraient être principalement dues à d'autres mécanismes d'échappement immunitaire. Dans l'analyse de survie, les caractéristiques composites intégrant les 21 voies de signalisation se sont avérées supérieures aux analyses de protéines individuelles pour la stratification des patients, démontrant ainsi pleinement l'intérêt d'une analyse combinée multiparamétrique.
Afin de garantir la fiabilité des résultats, toutes les principales conclusions ont été validées dans des cohortes indépendantes issues du TCGA. Malgré des différences significatives d'origine et de caractéristiques cliniques entre les deux populations, les principaux résultats sont restés très cohérents (coefficient de corrélation de Spearman de 0,88 au niveau du sous-type de cancer).Les 80 associations significatives communément identifiées ont montré un enrichissement statistique extrêmement élevé (p<2×10⁻⁹).Parallèlement, la population virtuelle de Providence Health a montré 331 associations significatives supplémentaires au niveau pan-cancer par rapport à TCGA, soulignant la valeur unique des données réelles à grande échelle.
L'analyse exploratoire a également révélé l'intérêt des motifs spatiaux complexes. Des indicateurs tels que l'entropie, le rapport signal/bruit et la netteté ont surpassé la simple densité d'activation dans respectivement 89, 63 et 79 paires protéine-biomarqueur. L'étude a également mis en évidence des effets synergiques entre les protéines.La combinaison de CD138 et CD68 a surpassé les protéines individuelles dans la prédiction de 20 biomarqueurs.Treize de ces différences étaient statistiquement significatives, suggérant que les plasmocytes et les macrophages pourraient travailler ensemble pour combattre les tumeurs par un mécanisme faisant intervenir des anticorps.
L'intelligence artificielle au service de la recherche sur le cancer : des cartes protéiques virtuelles aux nouvelles frontières de la recherche sur le cancer
La génération d'images de protéomique virtuelle à partir de lames histologiques de routine grâce à l'IA est au cœur de l'innovation en pathologie numérique et en biologie computationnelle. Cette voie a suscité l'intérêt des plus grandes institutions académiques du monde et a également stimulé les pratiques commerciales des entreprises de biotechnologie.
Dans le milieu universitaire,Le modèle HEX publié par l'université de Stanford dans Nature MedicineEntraîné sur 819 000 paires de blocs d'images, ce système peut prédire l'expression spatiale de 40 biomarqueurs, offrant une couverture protéique plus étendue que GigaTIME. Le système DeepHeme, publié par l'Université de Californie à San Francisco dans la revue Science Translational Medicine et basé sur près de 50 000 jeux de données multicentriques de haute qualité, a permis une classification précise de 23 types de cellules de moelle osseuse, proposant ainsi un modèle pour l'automatisation du diagnostic des maladies hématologiques.
Dans ce secteur, Reveal Biosciences bénéficie du soutien de la Fondation Bill & Melinda Gates.Développer une plateforme pour extraire des « biomarqueurs numériques » à partir d'images pathologiques.Accélérer la recherche en santé mondiale. Une autre approche consiste à réduire les coûts grâce à l'innovation matérielle, comme les dispositifs microfluidiques de Micronit qui diminuent considérablement la consommation d'échantillons et de réactifs. La plateforme de diagnostic des nodules pulmonaires d'Optellum, approuvée par la FDA, offre un modèle commercial et un précédent réglementaire pour l'extraction d'informations plus approfondies à partir de données de routine, en vue de la prise de décision clinique.
GigaTIME représente une étape importante dans ce domaine.Cela démontre non seulement l'énorme potentiel de l'IA multimodale dans l'étude du microenvironnement immunitaire tumoral, mais fournit également un cadre technique réutilisable et des ressources de données pour les recherches ultérieures.Les développements futurs dépendront de l'avancement combiné des capacités de génération de données en « réalité virtuelle » et des technologies de détection à faible coût, ce qui permettra à terme de disposer d'outils transformateurs pour comprendre la complexité des tumeurs et accélérer la médecine de précision.
Liens de référence :
1.https://mp.weixin.qq.com/s/AsqSemP3idCbIJ7xQ3gXGg
2.https://mp.weixin.qq.com/s/umg-UrMm6Qe-R-MbLpLZOQ








