Command Palette
Search for a command to run...
En Intégrant Des Données Sur Les Séquences Protéiques, Les Structures Tridimensionnelles Et Les Caractéristiques Fonctionnelles, Une Équipe Allemande a Construit Une « Vue Panoramique » Des Ligases d'ubiquitine E3 Humaines Basée Sur l'apprentissage métrique.
Chez les organismes, la dégradation et le renouvellement opportuns des protéines cellulaires sont essentiels au maintien de l'homéostasie protéique. Le système ubiquitine-protéasome (UPS) est un mécanisme central de régulation de la transduction du signal et de la dégradation des protéines. Au sein de ce système, les ligases E3 de l'ubiquitine, unités catalytiques clés, sont responsables de la reconnaissance de substrats spécifiques et de la catalyse de leur ubiquitine, régulant ainsi la dégradation, la localisation et l'état fonctionnel des protéines. De plus, les ligases E3 régulent également les voies immunitaires et inflammatoires. Du fait de leur expression tissulaire spécifique et de leur association avec des syndromes développementaux et métaboliques (dont la progression du cancer), les ligases E3 sont devenues des cibles thérapeutiques prometteuses, notamment pour des cibles auparavant difficiles à atteindre.
Comparativement aux enzymes E1 (environ 10 types) et E2 (environ 50 types), un grand nombre de ligases E3 humaines (environ 600 types) ont été identifiées. Néanmoins, de nombreuses ligases E3 humaines ne sont encore que partiellement caractérisées, et un grand nombre restent hypothétiques ou inconnues. À ce jour,Les ligases E3 étudiées présentent une forte hétérogénéité.Cela en fait l'une des classes d'enzymes les plus diversifiées, ce qui constitue un obstacle majeur à la reconnaissance de motifs et à la recherche à grande échelle. Par conséquent, une caractérisation et une analyse détaillées du génome des ligases E3 humaines — l'ensemble des ligases E3 codées par le génome humain — sont essentielles à une compréhension globale de leurs fonctions biologiques.
Dans ce contexte,Une équipe de recherche de l'université Goethe en Allemagne a classifié le « ligome E3 humain ».Il intègre des données à plusieurs niveaux, notamment les séquences protéiques, la composition des domaines, la structure tridimensionnelle, la fonction et les profils d'expression.La méthode de classification de l'équipe est basée sur le paradigme d'apprentissage métrique et utilise un cadre hiérarchique faiblement supervisé pour capturer les véritables relations entre la famille E3 et ses sous-familles.Cette approche élargit la classification traditionnelle des enzymes E3 (classes RING, HECT et RBR), fait la distinction entre les complexes multi-sous-unités et les enzymes monomériques, et associe les enzymes E3 à des substrats et à des cibles médicamenteuses potentielles.
Les résultats de cette recherche, intitulée « Une classification multi-échelle décode la complexité du ligome E3 humain », ont été publiés dans Nature Communications.
Points saillants de la recherche :
* Cartographier l'architecture du domaine, la structure tridimensionnelle, la fonction, le réseau de substrats et les interactions avec les petites molécules des ligases E3 existantes dans un cadre de classification afin d'obtenir des informations générales et spécifiques à la famille.
* Le cadre de classification multi-échelle développé couvre à la fois les mécanismes E3 typiques et atypiques, fournissant une feuille de route complète pour comprendre le vaste paysage biologique des ligases E3.
* Ouvre de nouvelles perspectives pour le développement de stratégies d'intervention médicamenteuse basées sur les réseaux E3-substrat.

Adresse du document :
https://www.nature.com/articles/s41467-025-67450-9
Suivez notre compte WeChat officiel et répondez « enzyme E3 » en arrière-plan pour obtenir le PDF complet.
Autres articles sur les frontières de l'IA :
https://hyper.ai/papers
Jeu de données : Construction de données sur l’E3 ubiquitine ligase humaine
L'équipe de recherche a d'abord intégré les données de l'ubiquitine ligase E3 humaine provenant de huit sources de données indépendantes.En incluant les publications antérieures et les bases de données publiques (E3Net, UbiHub, UbiNet 2.0, UniProt, BioGRID, etc.), un jeu de données préliminaire de 1 448 protéines a été constitué. Les doublons et les faux positifs potentiels ont été éliminés par comparaison croisée et évaluation de la cohérence des données issues de diverses sources. Par la suite, grâce aux caractéristiques des domaines catalytiques RING, HECT et RBR fournies par InterPro, 462 ligases E3 d'ubiquitine catalytiques à haute fiabilité ont été sélectionnées, constituant ainsi le génome final des ligases E3 humaines.
Dans les complexes E3 multi-sous-unités (tels que les ligases Cullin-RING), trois sous-unités fonctionnellement distinctes (protéine d'échafaudage, protéine aptamère et protéine réceptrice) agissent de concert pour localiser les molécules E2~Ub sur des substrats spécifiques. Les protéines d'échafaudage, volumineuses, rigides et situées au centre du complexe (comme la famille des Cullines, Cul1 à Cul5), organisent l'ensemble du complexe ligase en se liant simultanément aux sites d'ancrage de la sous-unité catalytique à domaine RING et de l'aptamère/récepteur. La protéine aptamère assure la liaison entre les modules, connectant la face d'ancrage N-terminale de la protéine d'échafaudage aux récepteurs de substrat individuels. La protéine réceptrice détermine la spécificité du substrat en reconnaissant et en se liant directement aux signaux de dégradation (dégrons) présents sur le substrat afin de déterminer quels substrats seront ubiquitinisés (par exemple, Skp2, Keap1, VHL).L'équipe de recherche a annoté et catégorisé indépendamment trois sous-unités : 151 aptamères, 106 récepteurs et 8 protéines d'échafaudage.Ils ont également utilisé leurs interactions protéine-protéine (IPP) pour cartographier les substrats de l'E3 multi-sous-unité.
Par la suite, lors de l'étape de sélection du domaine catalytique, les chercheurs ont utilisé la capacité catalytique comme critère principal pour filtrer rigoureusement les protéines candidates.En utilisant des bases de données de domaines telles qu'InterPro, le système a identifié des domaines catalytiques clés directement liés à l'activité E3, notamment RING, HECT et RBR.Seules les protéines contenant explicitement ces domaines et assurant leur fonction de ligation de l'ubiquitine aux niveaux de la séquence et de la structure sont conservées pour la construction de la ligase E3 catalytique finale. Ce processus élimine efficacement les protéines accessoires qui participent uniquement à la régulation sans posséder d'activité catalytique directe, garantissant ainsi la cohérence fonctionnelle du complexe E3 central.
Un cadre de classification multi-échelle basé sur l'apprentissage métrique
Pour saisir les relations complexes au sein du génome de la ligase E3 humaine,Des chercheurs ont utilisé des méthodes d'apprentissage automatique pour apprendre une métrique de distance émergente.La structure générale est illustrée dans le diagramme suivant :
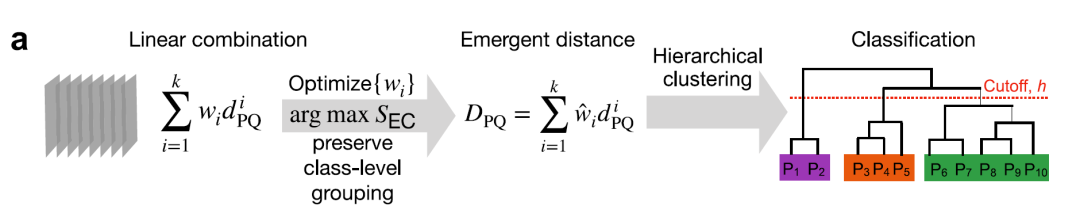
① Mesure de distance multi-échelle
Les chercheurs ont codé les relations par paires entre les ligases E3 en calculant 12 distances différentes.Ces distances couvrent plusieurs niveaux de granularité : séquence primaire, architecture du domaine, structure tertiaire, fonction, localisation subcellulaire et expression dans la lignée cellulaire/le tissu.Toutes les mesures de distance sont mises à l'échelle sur l'intervalle [0,1] pour la comparaison et la combinaison, comme indiqué dans la figure ci-dessous :
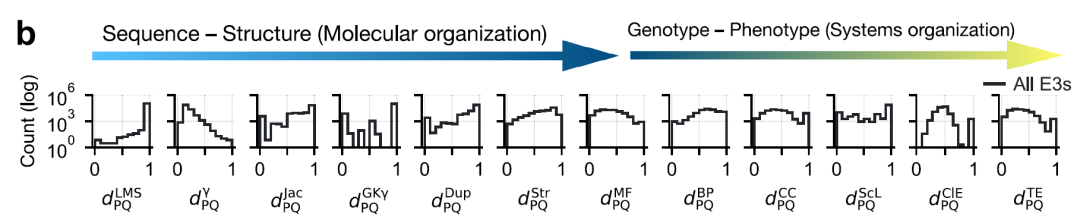
* Niveau de séquence : La distance de score de correspondance locale (LMS) (sans correspondance par paires) et la distance γ basée sur l'alignement ont été utilisées.
* Au niveau de l'architecture du domaine : trois distances ont été calculées : la distance de Jaccard, la distance γ de Goodman-Kruskal et la distance de répétition du domaine.
* Niveau structurel 3D : utilisation du score TM du modèle AlphaFold2
* Niveau fonctionnel : La distance fonctionnelle entre les protéines et P et Q est mesurée à l'aide de la similarité sémantique des annotations GO, couvrant trois ontologies : * Fonction moléculaire (MF), processus biologique (BP) et composant cellulaire (CC).
* Distance de localisation subcellulaire
* Distance de co-expression entre les tissus et les lignées cellulaires
② Optimisation des métriques, regroupement, rééchantillonnage et classification
Les quatre distances principales (γ, Jaccard, structure et fonction moléculaire) sont pondérées et intégrées, les poids étant optimisés par un apprentissage faiblement supervisé et l'indice de similarité du centre des éléments (SEC), comme indiqué dans la figure ci-dessous, pour obtenir l'indice combiné optimal.
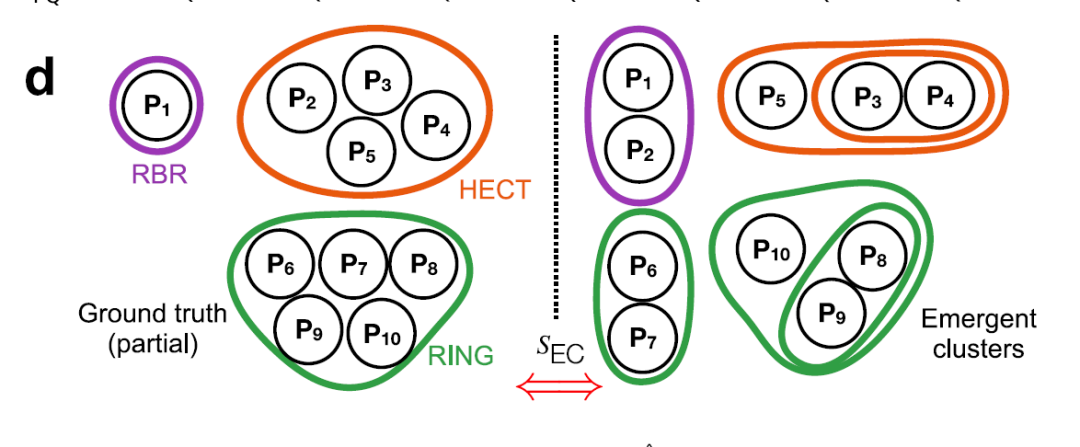
Un regroupement hiérarchique a été effectué en utilisant la méthode de variance minimale de Ward.Le support est calculé par la méthode de rééchantillonnage bootstrap pour générer le dendrogramme E3 final. Les clusters émergents optimaux sont obtenus avec un seuil de découpage d'arbre h = 0,25, divisant systématiquement les 462 clusters E3 en 13 familles : 10 familles RING, 2 familles HECT et 1 famille RBR, comme illustré dans la figure ci-dessous.
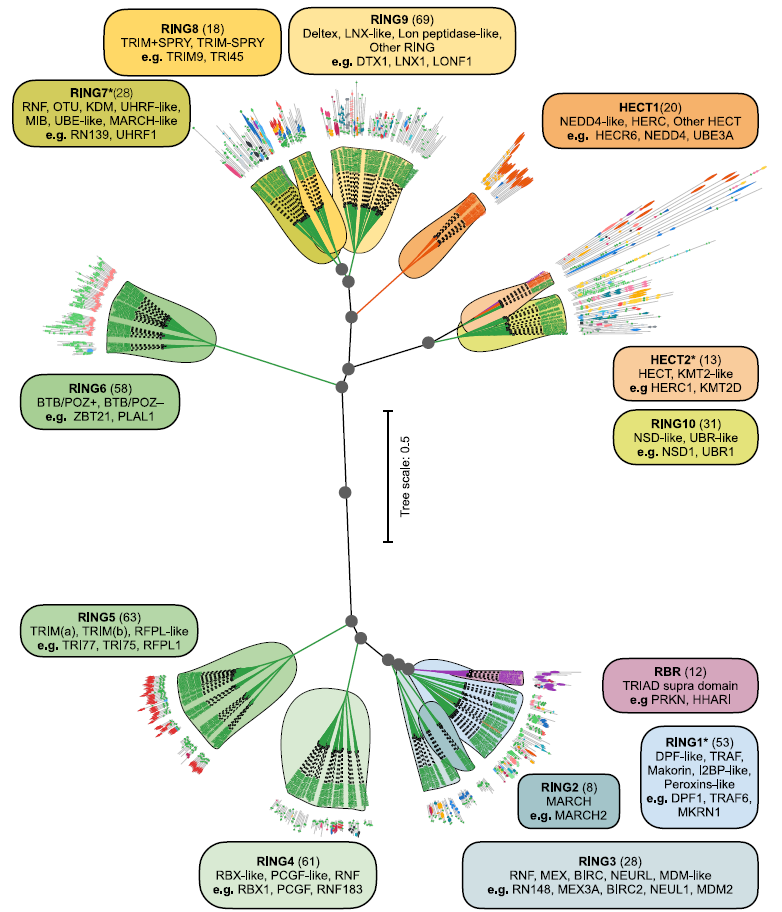
Chaque famille fait l'objet d'une analyse manuelle plus poussée des caractéristiques de séquence et de domaine afin d'identifier les sous-familles et les protéines aberrantes.
③ Probabilité de regroupement et de liaison des petites molécules
Projection UMAP 2D intégrée pour le regroupement de petites moléculesVingt petits groupes moléculaires représentatifs ont été identifiés en combinant les pics de densité locale.La probabilité que chaque cluster se lie à la protéine E3 a été quantifiée par des propensions transformées logarithmiquement (LPij), fournissant des indications pour le développement ultérieur de PROTAC et la conception ciblée de petites molécules.
Une évaluation détaillée de l'intégrité du génome de la ligase E3 humaine a été fournie.
① Organisation détaillée du génome de la ligase E3 humaine
Pour pallier les difficultés liées à la diversité des stratégies et aux définitions parfois incohérentes employées dans les études existantes concernant l'organisation du système E3, cette équipe de recherche a défini explicitement les composants catalytiques de ce système comme des séquences polypeptidiques contenant un ou plusieurs domaines catalytiques. Ce standard objectif permet une annotation précise et une analyse ciblée du système E3.Au final, les chercheurs ont découvert que 462 séquences polypeptidiques de l'ensemble des données contenaient au moins un domaine catalytique.Ces polypeptides constituent un génome de ligase E3 humaine finement organisé, comme le montre la figure ci-dessous :
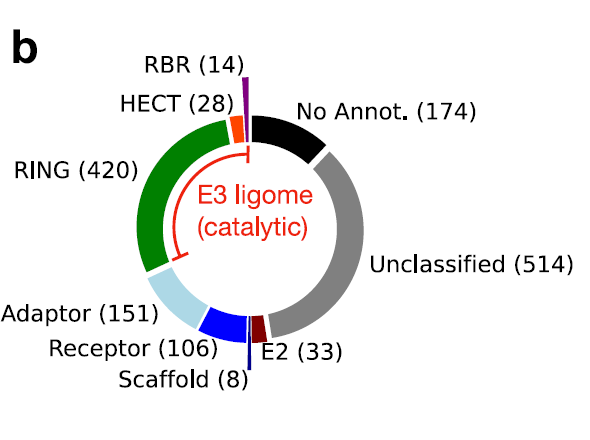
Pour vérifier la fiabilité du processus de tri, les chercheurs ont défini un score de consensus pour chaque protéine en fonction de sa fréquence d'apparition dans des ensembles de données provenant de différentes sources.Les résultats ont montré que les ligases E3 de classe HECT et RBR étaient très cohérentes dans l'ensemble de données (score de consensus ≥ 0,6, barres orange et violettes).Les scores de consensus pour la classe RING (barres vertes) sont largement distribués, ce qui indique une difficulté d'annotation, comme le montre la figure ci-dessous :
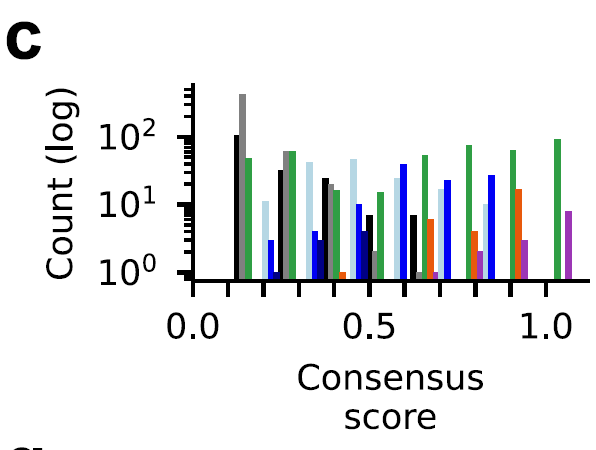
En utilisant cette méthode, les chercheurs ont minimisé les faux positifs et les vrais négatifs, inclus des E3 catalytiquement actifs hautement fiables et pris en compte les pseudo-E3 et d'autres E3 dont l'activité catalytique n'avait pas été vérifiée, fournissant ainsi une évaluation détaillée de l'intégrité du génome de la ligase E3 humaine.
② Différenciation fonctionnelle de la ligase E3 humaine
Pour évaluer la fonction de la ligase E3 humaine, les chercheurs ont recherché des délétions par CRISPR-Cas9 dans le gène UPS, en utilisant la viabilité cellulaire comme phénotype principal. Les résultats ont montré que…Au total, 53 composants E3 catalytiques et 32 composants E3 non catalytiques ont été identifiés comme étant cruciaux pour la viabilité cellulaire.Comme indiqué ci-dessous :
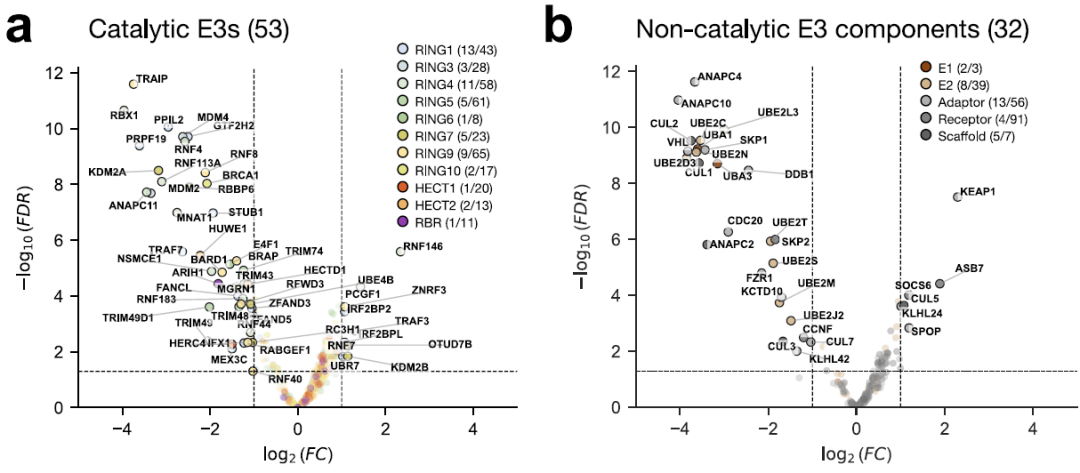
L'analyse GO de 53 E3 clés a révélé un enrichissement significatif en composants nucléaires et en processus de dommages à l'ADN, de réplication et de réparation, comme illustré dans la figure ci-dessous, soulignant leur rôle central dans le maintien de l'intégrité du génome et la régulation nucléaire. Ces résultats mettent en évidence les composants E3 essentiels à la survie cellulaire.
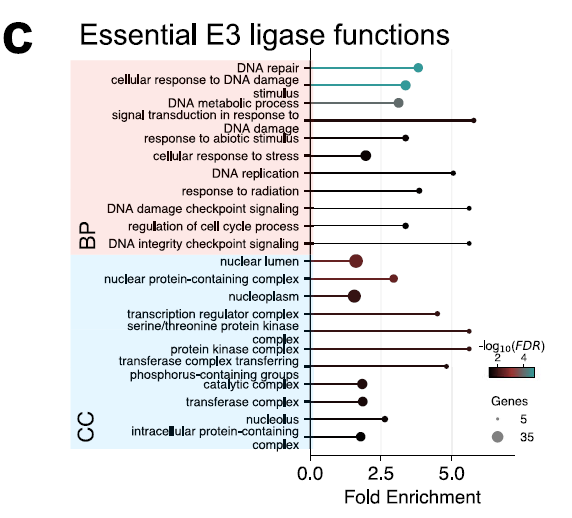
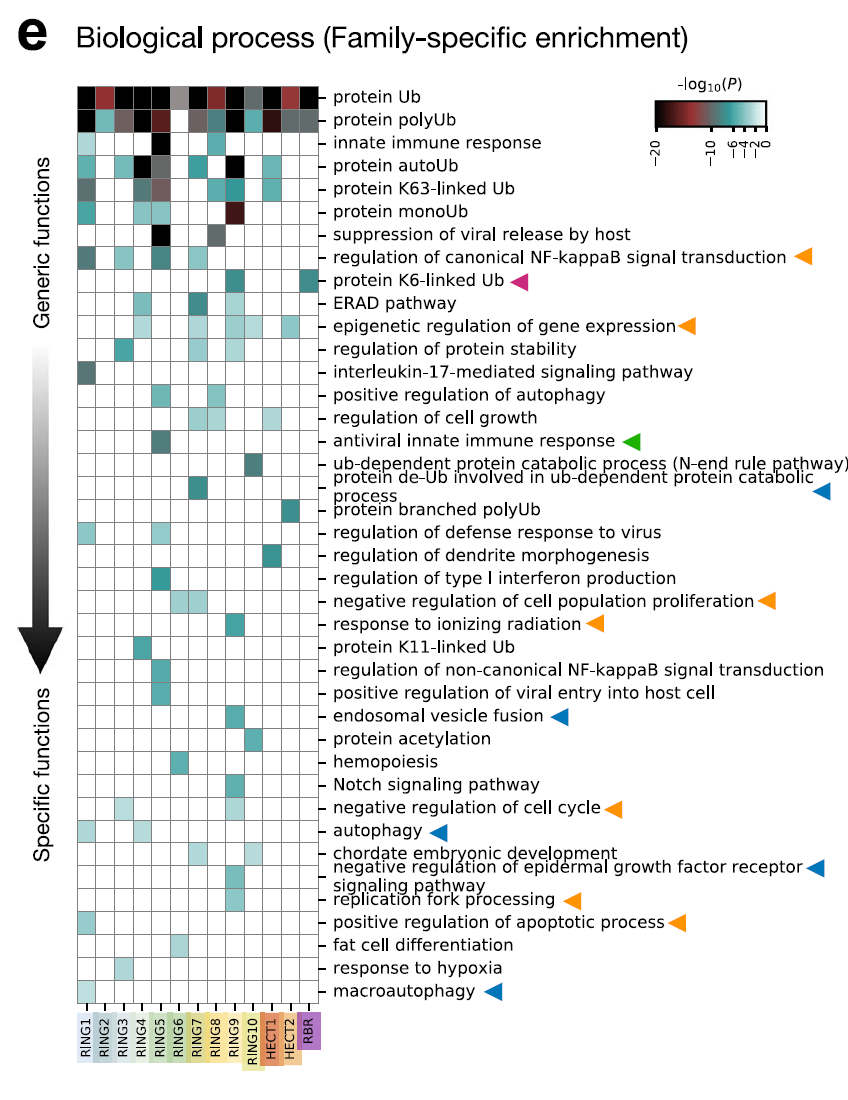
Une analyse d'enrichissement GO a été réalisée sur 13 familles E3 à l'aide de Metascape, et le réseau a été visualisé avec Cytoscape. Les résultats montrent que…Les différentes familles jouent des rôles distincts dans la sélection du substrat, la localisation cellulaire et la fonction catalytique.Comme illustré dans la figure ci-dessous. Par exemple, les membres de la famille RBR, RNF14, RNF144A et PRKN, sont spécifiques de l'ubiquitine liée à la lysine 6 (K6). La chaîne liée à K6 peut marquer les complexes ARN-protéines bloqués (RNF14), l'adaptateur de détection de l'ADN STING (RNF144A) pour activer la signalisation de l'interféron, et les mitochondries endommagées pour leur élimination (PRKN). De même,Les TRIM E3 (RING5) sont significativement enrichis dans les réponses immunitaires innées antivirales et régulent l'activité des récepteurs de reconnaissance de motifs dans les cellules.Par exemple, les réactions médiées par RIG-1 et MDA5.
④ Carte de la capacité des ligases E3 humaines à devenir des cibles thérapeutiques
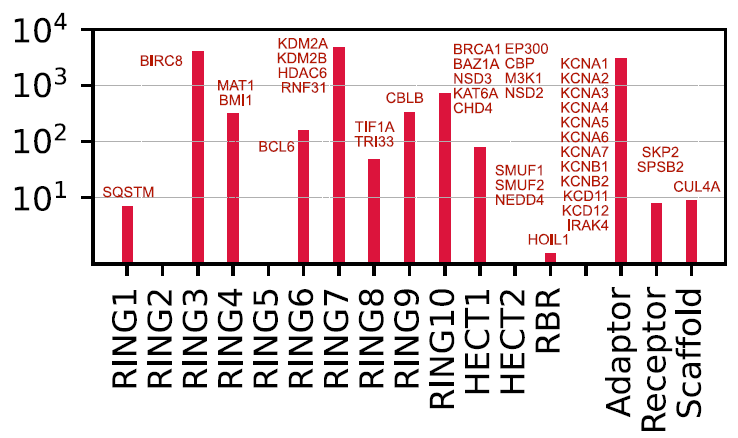
Afin d'explorer des voies thérapeutiques potentielles basées sur une action proximale, les chercheurs ont cartographié les opérandes E3 existants, dérivés de chimères de ciblage de la dégradation protéique (PROTAC) et de ligands E3 connus, sur différentes E3 et leurs familles. Actuellement, seules 16 protéines (9 E3 catalytiques et 7 adaptateurs) peuvent être ciblées directement par les opérandes E3 existants. La plupart des opérandes E3 conçus ciblent des protéines adaptatrices (telles que VHL et CRBN), tandis qu'un très petit nombre seulement cible directement les E3 catalytiques (telles que XIAP, MDM2/4/7 et BIRC2/3/7).
L'analyse des plus proches voisins utilisant la ligase E3 humaine de cette étude a révélé cinq protéines fortement corrélées (BIRC8, RN166/181/141 et UBR2).Comme indiqué ci-dessous :
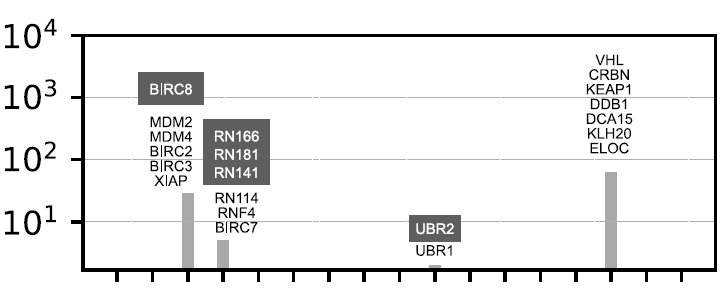
En raison de leur grande similarité structurelle (souvent des protéines homologues), les opérandes E3 existants peuvent être réutilisés pour cibler ces protéines.La cartographie des liants E3 à petites molécules a donné aux chercheurs accès à un ensemble potentiel de composés pouvant cibler 25 molécules E3 supplémentaires et 15 composants non catalytiques.Cette découverte révèle des cibles inexploitées, offrant une voie pour la conception rationnelle de composés de tête pour l'opérande E3, comme le montre la figure ci-dessous :
Les cadres multi-échelles offrent des outils puissants pour l'analyse des systèmes biologiques complexes.
En apprentissage automatique, un cadre multi-échelle désigne une méthode de modélisation ou une stratégie d'analyse capable de traiter des données à différents niveaux d'abstraction ou à différentes échelles de caractéristiques. Il ne s'agit pas d'un algorithme figé, mais d'un concept de conception visant à intégrer des informations locales et globales, ainsi que des caractéristiques à gros et à petits grains, améliorant ainsi l'expressivité et la capacité de généralisation du modèle.
L'intérêt de ce cadre de classification multi-échelle ne se limite pas à l'analyse systématique de la famille des ligases E3 ; son principal atout réside dans la fourniture d'un paradigme transférable et adaptable à grande échelle pour l'intégration des données omiques. Cette approche d'intégration inter-échelle permet naturellement son extension à d'autres données omiques multimodales, offrant ainsi un outil universel pour l'analyse systématique des systèmes biologiques complexes.
Par exemple, la cellule est l'unité fondamentale de la vie, et sa fonction et son devenir sont déterminés par des réseaux moléculaires complexes. Si les méthodes d'apprentissage profond classiques sont performantes pour l'identification des types cellulaires à partir de données de transcriptome unicellulaire, elles manquent d'interprétabilité biologique. Le 20 octobre 2025, des chercheurs du Centre national des sciences des protéines de Chine (Pékin) et de l'université Tsinghua ont proposé Cell Decoder, un cadre d'apprentissage profond interprétable à plusieurs échelles qui intègre des connaissances biologiques préalables.Elle permet une caractérisation hiérarchique et un raisonnement allant des gènes et des voies métaboliques aux processus biologiques, offrant une nouvelle approche pour décoder les types cellulaires au niveau de la cellule unique. Cell Decoder construit un graphe de connaissances biologiques multi-échelle en intégrant des réseaux d'interaction protéique, des correspondances gène-voie et des relations hiérarchiques de voies dans une architecture de réseau neuronal graphique.L'équipe de recherche a évalué de manière systématique Cell Decoder par rapport à neuf méthodes courantes sur des échantillons humains et murins provenant de sept jeux de données monocellulaires accessibles au public. Les résultats ont montré que Cell Decoder se classait premier en termes de précision de prédiction (0,87) et de score F1 macro (0,81), et conservait des performances stables même dans des conditions complexes telles que des perturbations dues au bruit, un déséquilibre des types cellulaires et des variations de distribution entre les lots.
Titre de l'article :Décodeur cellulaire : décodage de l’identité cellulaire grâce à un apprentissage profond explicable multi-échelle
Adresse du document :
https://link.springer.com/article/10.1186/s13059-025-03832-y
À plus long terme, ce cadre multi-échelle peut être davantage intégré aux données de protéomique spatiale, aux chimiothèques et aux informations spatiales chimiques, levant ainsi les barrières entre la recherche biologique fondamentale, l'analyse des mécanismes pathologiques et les applications translationnelles. Avec l'accumulation continue de données multi-omiques, ce cadre devrait jouer un rôle de plus en plus important dans la recherche en sciences de la vie et l'innovation biomédicale.
Références :
1.https://www.nature.com/articles/s41467-025-67450-9
2.https://blog.csdn.net/qazplm12_3/article/details/153948711
3.https://link.springer.com/article/10.1186/s13059-025-03832-y








