Command Palette
Search for a command to run...
Durch Die Zusammenarbeit Von MIT Und Harvard Wird Ein Hochselektives Substratdesign Erreicht Und Neuartige Protease-Spaltungsmuster Mithilfe Generativer KI entdeckt.

Im komplexen Netzwerk biochemischer Reaktionen lebender Organismen können Proteasen spezifisch Peptidbindungen spalten und dadurch eine Reihe wichtiger Lebensprozesse präzise regulieren – von der Blutgerinnung und Gewebereparatur über Immunreaktionen bis hin zur Krebsentstehung. Funktionsstörungen dieser Proteasen führen häufig direkt zum Auftreten und Fortschreiten verschiedener schwerwiegender Erkrankungen. Daher ist die Aufklärung der Wirkungsmechanismen von Proteasen und die präzise Regulierung ihrer Aktivität nicht nur eine zentrale Frage der Grundlagenforschung in den Lebenswissenschaften, sondern auch ein entscheidender Durchbruch für die Entwicklung neuartiger Diagnose- und Therapieverfahren.
Der Schlüssel zur Erreichung dieses Ziels,Der Schlüssel liegt darin, Peptidsubstrate zu finden, die hochgradig "passend" sind.Sie können als molekulare Sonden zur Verfolgung der Enzymaktivität, als Inhibitoren zur Blockierung abnormaler Aktivität oder sogar als „bedingte Aktivierungsschalter“ in Arzneimittelverabreichungssystemen zur Erreichung einer gezielten Therapie eingesetzt werden.
Die Entwicklung von Peptidsubstraten, die sowohl schnell von Zielproteasen gespalten werden als auch hochselektiv sind (nur von diesem Enzym erkannt werden und Kreuzreaktionen mit anderen Proteasen vermeiden), stellt die Wissenschaft seit jeher vor große Herausforderungen. Dieses Problem rührt von den komplexen biochemischen Wechselwirkungen zwischen Proteasen und Substraten her: Um sich an vielfältige physiologische Funktionen anzupassen, haben Proteasen breite Spaltungsspezifitäten entwickelt, und ihre aktiven Zentren müssen präzise an Peptidsubstrate (typischerweise etwa zehn Aminosäuren lang) binden. Selbst bei Betrachtung synthetischer Peptide mit nur zehn Aminosäuren, die die 20 häufigsten natürlichen Aminosäuren verwenden, ergeben sich theoretisch etwa 20¹⁰ (fast 10¹³) Sequenzkombinationen, wodurch sich ein nahezu unendlicher Raum für weitere Untersuchungen eröffnet. Erschwerend kommt hinzu, dass…Proteasen mit ähnlichen Funktionen stammen oft von einem gemeinsamen Vorfahren ab und weisen ähnliche Strukturen im aktiven Zentrum auf, wodurch sie sehr anfällig für „Kreuzerkennung“ sind.Dies macht es besonders schwierig, aus einer Vielzahl von Möglichkeiten hochspezifische Substrate auszuwählen.
Um diesen Engpass zu überwinden, haben Forscher zahlreiche Versuche unternommen. Traditionelle Methoden basieren oft auf bekannten Spaltstellen oder enzymatischen Informationen natürlicher Proteine, was zu geringer Effizienz und Schwierigkeiten bei der Gewinnung idealer künstlicher Substrate führt. Rationales Design auf Basis chemisch-biologischer Erkenntnisse ist in der Regel aufwendig, hat einen begrenzten Durchsatz und konzentriert sich meist auf einzelne Proteasen, was eine Skalierung erschwert. Obwohl Hochdurchsatz-Screening-Technologien in den letzten Jahren die Effizienz etwas verbessert haben, weisen sie weiterhin Einschränkungen wie komplexe Handhabung und hohe Kosten auf.Die meisten bestehenden rechnergestützten Vorhersagemethoden können lediglich bestimmen, „ob geschnitten werden soll“, und sind nicht in der Lage, die Schneidleistung genau zu bestimmen. Daher werden sie den Anforderungen der eingehenden Mechanismusforschung und der technischen Anwendungen nicht gerecht.
In diesem ZusammenhangDas MIT und die Harvard University haben gemeinsam CleaveNet vorgeschlagen, einen durchgängigen Designprozess auf Basis künstlicher Intelligenz.Durch die Synergie zwischen prädiktiven und generativen Modellen zielt dieser Ansatz darauf ab, das bestehende Paradigma des Proteasesubstratdesigns zu revolutionieren und völlig neue Lösungen für die damit verbundene Grundlagenforschung und biomedizinische Entwicklung bereitzustellen.

Papieradresse:
https://www.nature.com/articles/s41467-025-67226-1
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „CleaveNet“, um das vollständige PDF zu erhalten.
Weitere Artikel zu den Grenzen der KI:
Szenarioübergreifende Validierung, die die Generalisierungsfähigkeit des CleaveNet-Modells stärkt, unterstützt durch Datensätze aus mehreren experimentellen Szenarien.
Bei der Entwicklung und Validierung des CleaveNet-Modells wurden in dieser Studie zwei Datensätze integriert, die sich hinsichtlich Sequenzzusammensetzung und experimenteller Methoden deutlich unterschieden, um die Zuverlässigkeit und Generalisierungsfähigkeit des Modells zu gewährleisten.
Der von den Forschern verwendete Kerndatensatz stammt aus einer veröffentlichten Studie, in der die Spaltungsaktivität einer Substratbibliothek mit etwa 18.500 synthetischen Dekapeptiden gegenüber 18 Matrix-Metalloproteinasen (MMPs) mithilfe der mRNA-Display-Technologie systematisch charakterisiert wurde.Jeder Substrat-Protease-Kombination ist ein standardisierter Spaltungseffizienzwert (Zₛₘ) zugeordnet, der die relative Spaltungsintensität quantifiziert.
Um die Strenge der Bewertung weiter zu gewährleisten und eine Überschätzung aufgrund von Sequenzähnlichkeit zu vermeiden,Die Forscher führten eine Homologiefilterung am ursprünglichen Testdatensatz durch:Die Forscher berechneten den minimalen Levenstein-Abstand zwischen jeder Testsequenz und allen Sequenzen des Trainingsdatensatzes und entfernten 816 Sequenzen mit einem Abstand von weniger als 3, die dem Trainingsdatensatz sehr ähnlich waren. Schließlich erhielten sie einen „mRNA-Display-Testdatensatz“ mit 2.901 nicht überlappenden Sequenzen. Dieser Teildatensatz wurde in keiner Phase des Modelltrainings verwendet und diente ausschließlich der internen Leistungsvalidierung.
Um die Anpassungsfähigkeit des Modells bei drastisch unterschiedlichen biochemischen Rahmenbedingungen unabhängig zu testen,Die Studie führte außerdem einen völlig unabhängigen, außerhalb der regulären Datenverteilung liegenden Datensatz ein, der als „Fluoreszenz-Testset“ bezeichnet wird.Dieser Datensatz umfasst 71 synthetische Peptide unterschiedlicher Länge (7–14 Aminosäuren), deren Spaltungsaktivität für sieben rekombinante MMP-Proteine mittels klassischer In-vitro-Experimente auf Basis von Fluoreszenzresonanzenergietransfer (FRET) validiert wurde. Dieser Datensatz unterscheidet sich grundlegend vom Kerndatensatz, der mittels mRNA-Display-Technologie generiert wurde, hinsichtlich der Peptidlängenverteilung, der Aminosäurezusammensetzung und vor allem der experimentellen Detektionsprinzipien. Dieses gezielte Design dient als entscheidender Vergleichsmaßstab zur Bewertung der Fähigkeit des CleaveNet-Modells, über spezifische experimentelle Bedingungen hinaus universelle biochemische Muster zu erfassen.
CleaveNet prognostiziert und generiert kollaborative geschlossene Regelkreise.
Wie in der folgenden Abbildung dargestellt, besteht der Kern von CleaveNet aus zwei sich ergänzenden und kollaborativen Rechenmodulen: dem Vorhersagemodul (CleaveNet Predictor) und dem Generierungsmodul (CleaveNet Generator).Zusammen bilden sie einen vollständig geschlossenen Kreislauf aus Design und Evaluierung.
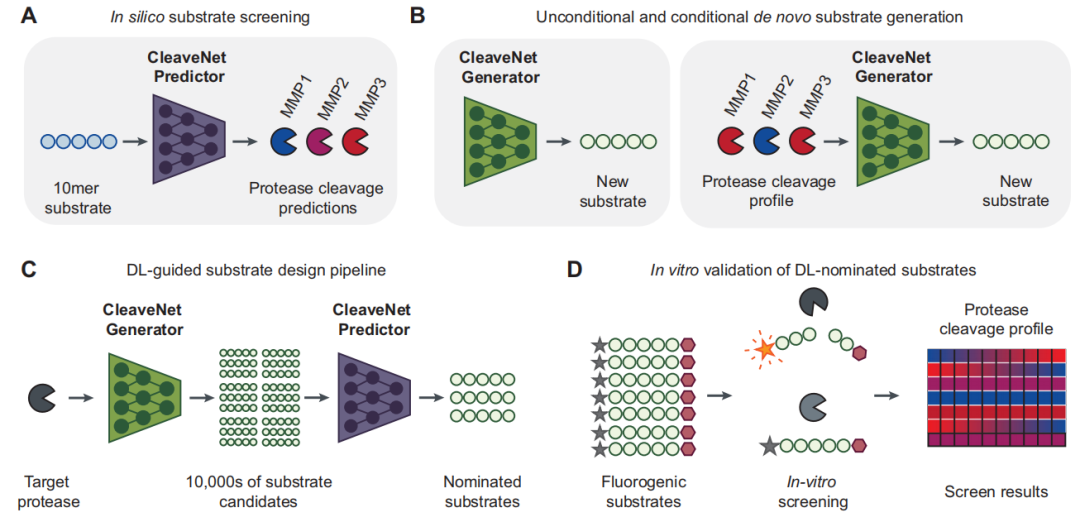
Das Vorhersagemodul hat zum Ziel, das Problem der schnellen und genauen Bewertung der Spaltungsaktivität von Kandidatensubstraten aus einem massiven Sequenzraum zu lösen.Die Forscher konstruierten es als Sequenzfunktionsregressionsmodell mit mehreren Ausgaben. Konkret nimmt das Modell eine Aminosäuresequenz als Eingabe und seine Kernaufgabe besteht darin, gleichzeitig den vorhergesagten Cut-Score (Ŵₛₘ) der Sequenz für alle 18 MMPs auszugeben und gleichzeitig die Unsicherheit (σₛₘ) jeder Vorhersage abzuschätzen.
Um eine höhere Vorhersagerobustheit zu erreichen, wurde in dieser Studie eine Modellensemble-Strategie angewendet:Fünf identische Vorhersagemodelle wurden unabhängig voneinander mit dem mRNA-Display-Trainingsdatensatz trainiert. Der finale Vorhersagewert ergab sich aus dem Mittelwert ihrer Ausgaben. Die Unsicherheit der Vorhersagen wurde durch die Standardabweichung dieser fünf Ergebnisse quantifiziert. Durch die Festlegung eines anpassbaren Schwellenwerts (Zₜ) kann das Modell kontinuierliche Vorhersagewerte zudem einfach in eine binäre Entscheidung („geschnitten“ oder „ungeschnitten“) umwandeln und eignet sich somit für verschiedene Screening-Szenarien.
Bei der Entwicklung des Vorhersagemodells verglich diese Studie systematisch zwei gängige Architekturen der Sequenzmodellierung: bidirektionale Long Short-Term Memory Networks (LSTM) und Transformer. Erstere eignen sich hervorragend zur Erfassung von Sequenzabhängigkeiten, während letztere mit ihrem Aufmerksamkeitsmechanismus Interaktionen zwischen Aminosäuren global modellieren können und derzeit die bevorzugte Methode zur Repräsentation von Proteinsprachen darstellen. Aufgrund ihres Potenzials, das anhand umfangreicherer und vielfältigerer Datensätze demonstriert wurde,Die Forscher wählten schließlich die Transformer-Architektur als Grundlage für den CleaveNet Predictor.
Ziel des Generierungsmoduls ist die automatisierte und intelligente Entwicklung von Kandidatensubstraten.In dieser Studie wurde ein generatives Modell auf Basis eines autoregressiven Transformers trainiert, wodurch es in der Lage war, die universellen MMP-Schnittpräferenzen, die dem Datensatz innewohnen, aus der mRNA-Repräsentation zu lernen.Dieses Modell kann ohne zusätzliche Eingabebedingungen eine große Anzahl neuartiger und plausibler Peptidsequenzen generieren.
Um den Wert generativer Modelle wissenschaftlich zu bewerten und nicht einfach nur Zufall zu reproduzieren, haben Forscher eine robuste Basismethode namens „standortunabhängige Kontrolle“ entwickelt.Bei dieser Methode wird lediglich die unabhängige Verteilung jeder Aminosäureposition in den Trainingsdaten gezählt und anschließend eine Zufallsstichprobe auf dieser Grundlage durchgeführt, um Sequenzen zu generieren.Durch den Vergleich der von CleaveNet generierten Sequenzen mit dieser Basissequenz über mehrere Dimensionen hinweg können wir die komplexen biochemischen Muster, die das Modell gelernt hat und die über einfache statistische Zusammenhänge hinausgehen, klar identifizieren.
Die enge Zusammenarbeit zwischen den Vorhersage- und Generierungsmodulen ermöglicht es den Forschern, zunächst eine vielfältige Kandidatenbibliothek zu erstellen und anschließend ein effizientes und genaues virtuelles Screening daran durchzuführen. Dadurch steht eine leistungsstarke Rechenmaschine für die nachfolgende experimentelle Überprüfung zur Verfügung.
CleaveNet ermöglicht eine selektive und präzise Steuerung.
Nach Abschluss der Modellkonstruktion wurde in dieser Studie eine mehrstufige und systematische experimentelle Überprüfung der Leistungsfähigkeit von CleaveNet durchgeführt. Die Ergebnisse demonstrierten vollumfänglich den herausragenden Wert dieses Prozesses in Bezug auf Vorhersagegenauigkeit, Generierungsrationalität und praktische Anwendungseffektivität.
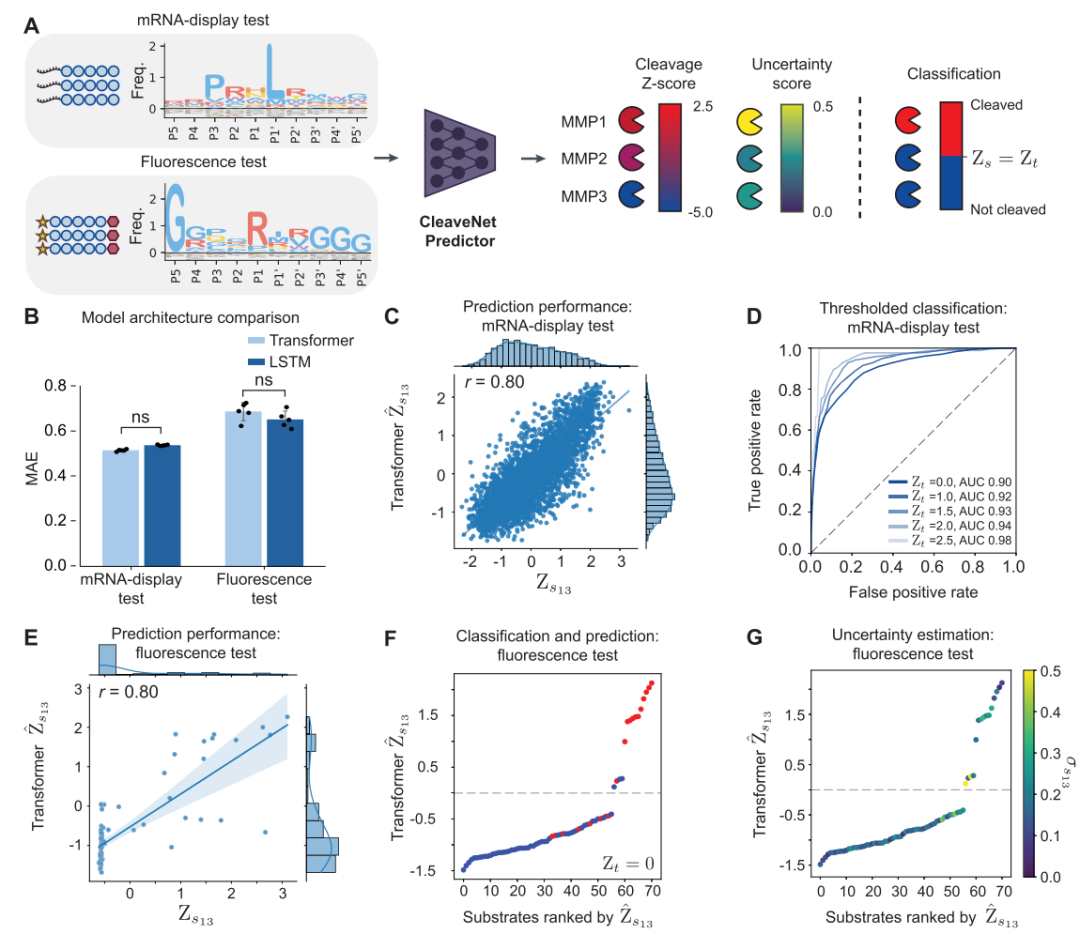
Erste,CleaveNet Predictor weist sowohl auf internen als auch auf externen Testdatensätzen hervorragende Vorhersagefähigkeiten auf.Auf einem Homologiefilter-Testdatensatz (mRNA-Display-Testdatensatz), der nie im Training verwendet wurde, zeigte der vom Modell vorhergesagte Wert (Ŵₛₘ) für MMP13 eine starke Korrelation mit dem experimentell gemessenen standardisierten Z-Wert (Zₛₘ) (Pearson-Korrelationskoeffizient r = 0,80). Die Leistung des Modells blieb ebenso robust, als die kontinuierlichen Vorhersagen in eine binäre Klassifizierung („Cut/Uncut“) umgewandelt wurden: Durch die Erstellung von ROC-Kurven (Receiver Operating Characteristic) und die Berechnung der Fläche unter der Kurve (AUC) stellten die Forscher fest, dass das Modell über verschiedene Entscheidungsschwellen hinweg eine hohe Diskriminierungsfähigkeit beibehielt, insbesondere bei der allgemein akzeptierten Cut-Schwelle (Zₜ = 2,5), wo die AUC 0,98 erreichte. Weitere strenge Tests wurden mit völlig unabhängigen Fluoreszenz-Testdatensätzen durchgeführt, die mit sehr unterschiedlichen experimentellen Methoden durchgeführt wurden.
Obwohl sich die Sequenzlänge, die Aminosäurezusammensetzung und das Detektionsprinzip dieses Datensatzes von denen der Trainingsdaten unterscheiden, weist der vom Modell vorhergesagte Cut-Score immer noch eine starke positive Korrelation mit dem experimentellen Wert auf (r = 0,80 für MMP13) und kann zwischen den experimentell verifizierten „geschnittenen“ und „ungeschnittenen“ Sequenzen genau unterscheiden.Dies bestätigt nachdrücklich, dass CleaveNet Predictor nicht nur Trainingsdatenmuster auswendig lernen kann, sondern auch die universellen biochemischen Gesetze erfasst, die die Substratspaltung durch Proteasen regeln.Es besitzt eine starke Generalisierungsfähigkeit.
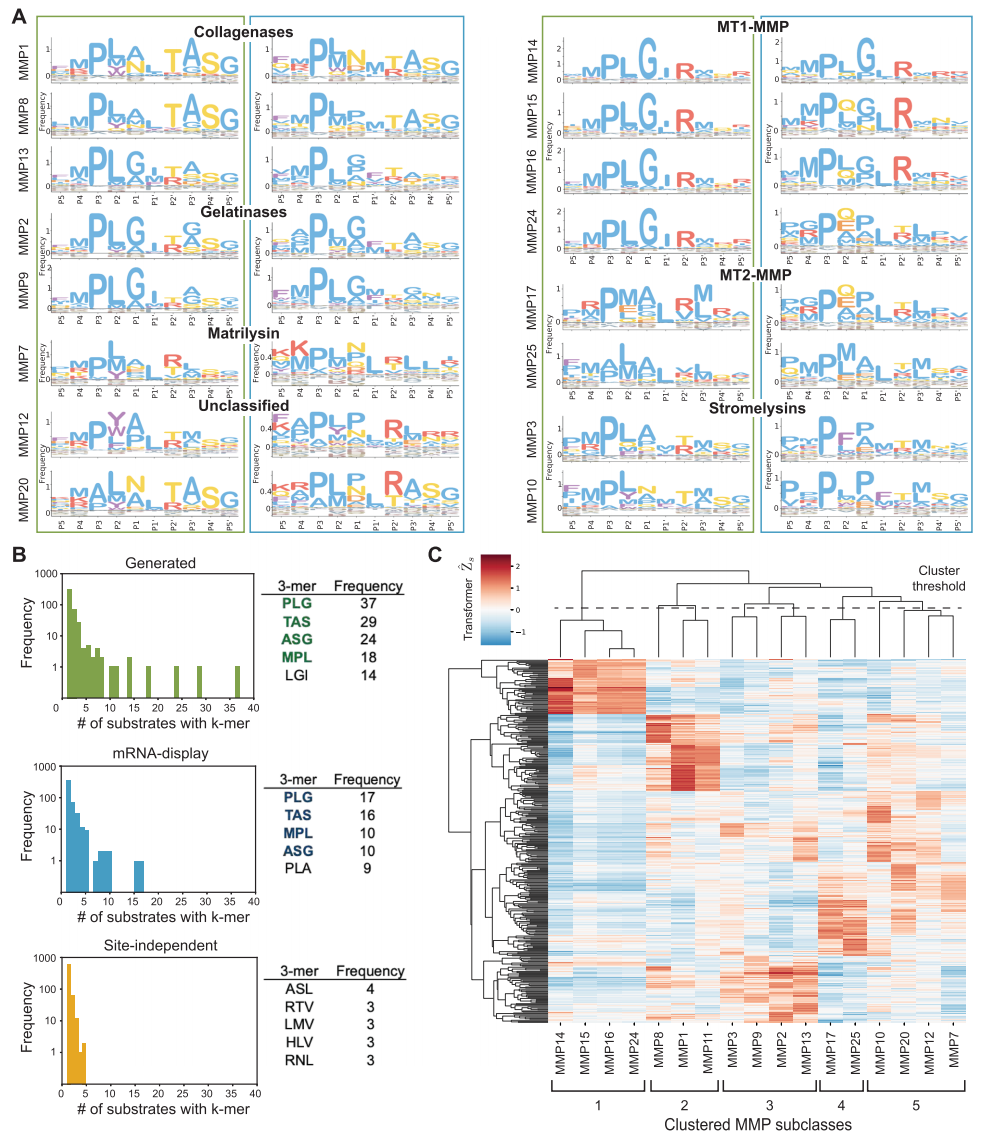
Zweitens,Die bioinformatische Analyse der von CleaveNet Generator erzeugten Sequenzen durch Forscher offenbarte dessen Funktionsweise und Neuartigkeit.Im Vergleich zu „ortsunabhängigen Kontrollsequenzen“, die ausschließlich auf zufälliger Stichprobenziehung der Häufigkeit einzelner Aminosäurepositionen basieren, reproduzieren die vom generativen Modell erzeugten Sequenzen die klassischen Spaltungsmotive der MMP-Familie genauer und weisen eine Aminosäureverteilung in wichtigen Substratbindungsstellen auf, die den tatsächlichen experimentellen Daten besser entspricht. Wichtiger noch,Die generierten Sequenzen stimmen hinsichtlich der allgemeinen biophysikalischen Eigenschaften (wie Hydrophobie und Ladung) mit dem realen Datensatz überein.Hochwertige Generierung bedeutet jedoch nicht einfach die Reproduktion der Trainingsdaten. Die Sequenzdiversitätsanalyse zeigte, dass der Anteil einzigartiger langer synthetischer Peptide, die sowohl in den generierten Sequenzen als auch im Trainingsdatensatz vorkommen, extrem gering war. Dies deutet darauf hin, dass das Modell eine Überanpassung vermied und neue Sequenzbereiche erschließen konnte, die nicht von den Trainingsdaten abgedeckt wurden.
Weiterführende funktionelle Clusteranalysen ergaben, dass die vorhergesagten Spaltungsaktivitätsspektren von Substraten mit hohen Punktzahlen, die von verschiedenen MMPs erzeugt wurden, auf der Grundlage der phylogenetischen Beziehungen der katalytischen Domänen der MMPs natürlich gruppiert werden konnten.Dies beweist, dass das generative Modell nicht nur die offensichtlichen Sequenzmuster erlernt, sondern auch intrinsisch Informationen über die funktionelle Differenzierung in der Evolution von Proteasen erfasst.Dies beweist die biologische Rationalität der erzielten Ergebnisse.
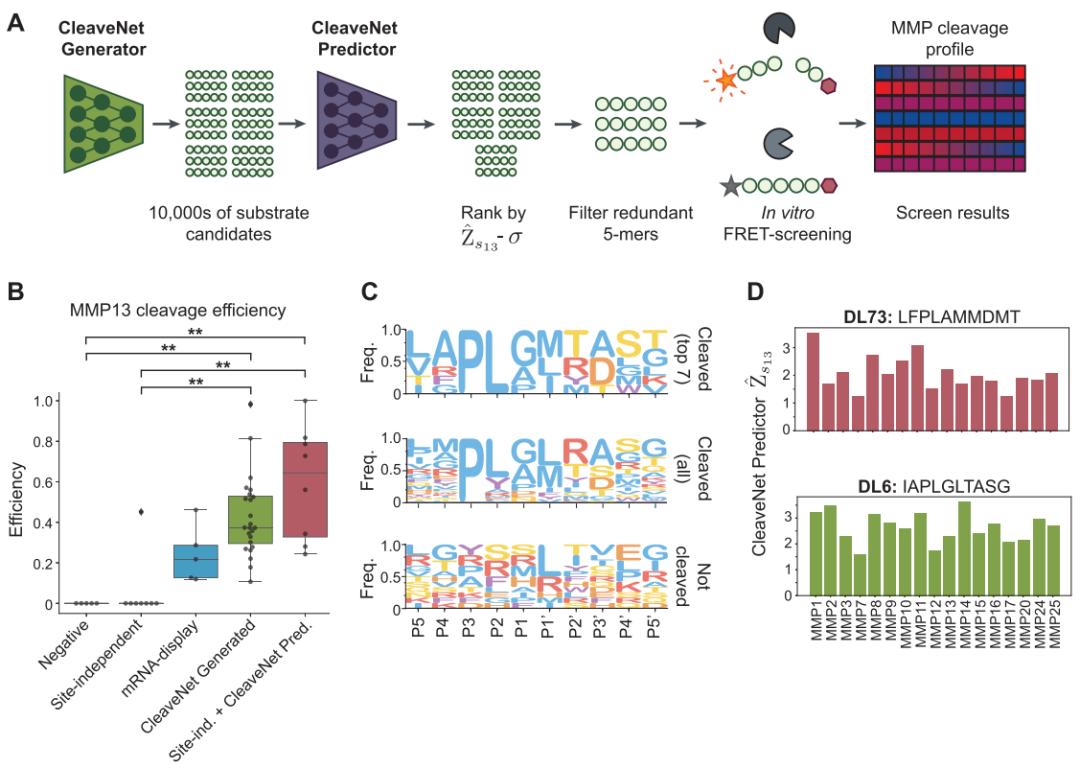
Die Gültigkeit aller computergestützten Designs wurde schließlich durch biochemische In-vitro-Experimente bestätigt. Die Forscher synthetisierten mehrere Sätze von Kandidatensubstraten, die mit CleaveNet gegen MMP13 gerichtet waren. Diese umfassten Sequenzen, die direkt vom generativen Modell generiert wurden, sowie Sequenzen, die vom prädiktiven Modell identifiziert wurden. FRET-Spaltungsversuche lieferten überzeugende Ergebnisse.Alle 24 Substrate, die mithilfe der CleaveNet-Pipeline entworfen wurden, konnten durch das rekonstruierte MMP13 erfolgreich gespalten werden, wobei eine Trefferquote von 100 % (TP3T) erreicht wurde.Darüber hinaus war die mittlere Schneidleistung signifikant höher als die der bekannten hocheffizienten Positivkontrollsubstrate im Trainingsdatensatz. Dies bestätigt die Fähigkeit dieses Verfahrens, hocheffiziente Substrate zu entwickeln.
Um das Potenzial des Verfahrens für anspruchsvollere Aufgaben, wie die Entwicklung hochselektiver Substrate, aufzuzeigen, wurde in dieser Studie eine Strategie der bedingten Generierung angewendet, wobei „hohe MMP13-Selektivität“ als Ziel im generativen Modell festgelegt wurde. Ein anschließendes groß angelegtes paralleles In-vitro-Screening (95 Substratpaare für 12 verschiedene MMPs) zeigte, dass die mittels bedingter Steuerung generierten Substrate eine hohe Selektivität aufwiesen.Die Spaltungsaktivität ist deutlich auf MMP13 ausgerichtet, was zu einer höheren Selektivität führt.
Besonders hervorzuheben ist, dass einige der entworfenen Substrate sowohl eine hohe Schneidleistung als auch eine hohe Selektivität aufweisen – eine hervorragende Kombination, die in den ursprünglichen Trainingsdaten äußerst selten ist und die die große Fähigkeit von CleaveNet unterstreicht, neue und qualitativ hochwertige Sequenzräume zu erkunden.
Zusammenfassend lässt sich sagen, dass CleaveNet mit seinen präzisen computergestützten Vorhersagen, der Generierung plausibler Sequenzen und der abschließenden experimentellen Verifizierung eine effiziente, zuverlässige und leistungsstarke Plattform für das Design von Proteasesubstraten geschaffen hat. Diese Forschung bietet nicht nur eine innovative KI-Lösung für die klassische Herausforderung der Proteaseaktivitätsregulation, sondern legt auch ein neues methodisches Fundament für zukünftige Forschung zur Proteasefunktion und die Entwicklung entsprechender Medikamente.
KI-gestützte Innovation im Design von Proteasesubstraten
Die KI-gestützte Substratdesign-Technologie von CleaveNet für Proteasen treibt weltweit Innovationen in den Bereichen Biowissenschaften und Biomedizin voran.
Das Team von David Baker an der University of Washington veröffentlichte bahnbrechende Forschungsergebnisse in der Fachzeitschrift Science.Zum ersten Mal wurde KI eingesetzt, um eine Serinhydrolase mit einem komplexen aktiven Zentrum von Grund auf zu entwerfen – eine der größten bekannten Enzymfamilien.Die Studie stellte ein neuartiges maschinelles Lernnetzwerk namens PLACER vor, das nicht nur erfolgreich ein aktives Enzym entwarf, das die Esterhydrolyse effizient katalysieren kann, sondern auch unerwartet fünf neue Proteinfaltungsmuster entdeckte und damit die strukturelle Vielfalt dieser Enzymfamilie erheblich erweiterte.
* Titel des Artikels: Computergestütztes Design von Serinhydrolasen
* Link zum Artikel:
https://www.science.org/doi/10.1126/science.adu2454
Darüber hinaus hat ein gemeinsames Team mehrerer europäischer Universitäten ein allgemeines Modell auf Basis der Transformer-Architektur entwickelt, das Protease-Substrat-Interaktionen präzise vorhersagen kann. Dieses Modell integriert globale, aus verschiedenen Quellen stammende Protease-Spaltungsdaten und ermöglicht so die effektive Vorhersage von Substratsequenzen über verschiedene Spezies hinweg. Seine Generalisierungsfähigkeit wurde in der Forschung an Proteasen verschiedener Krankheitserreger, darunter Bakterien und Viren, validiert und liefert eine wichtige Grundlage für das Sequenzdesign zur Entwicklung antiinfektiver Medikamente.
Es ist absehbar, dass sich mit der fortschreitenden Konvergenz von Computerbiologie, künstlicher Intelligenz und synthetischer Biologie das Design von Proteasesubstraten von einer Wissenschaft, die Kunst und Erfahrung vereint, zu einem hochgradig rationalisierten und systematisch entwickelten Forschungsfeld entwickeln wird. Dies wird nicht nur die Entwicklung neuartiger Medikamente, Diagnoseverfahren und umweltfreundlicher Biokatalysatoren beschleunigen, sondern uns potenziell auch helfen, die zugrundeliegende Logik der Lebensregulation zu entschlüsseln und so eine neue Ära der bedarfsgerechten Programmierung von Lebensfunktionen einzuleiten.








