Command Palette
Search for a command to run...
Um Die Vorhersagekraft Ungeordneter Proteinaggregate Neu Zu Gestalten, Veröffentlichen NVIDIA, MIT, Die Universität Oxford, Die Universität Kopenhagen, Peptone Und Andere Generative Modelle Und Neue Benchmarks.

In der Geschichte der Strukturbiologie galt der Grundsatz „Struktur bestimmt Funktion“ einst als nahezu unumstößliches Naturgesetz. Sowohl die klassische Helixstruktur des Insulins als auch die tetramere Struktur des Hämoglobins bestärkten die Auffassung, dass Proteine eine stabile dreidimensionale Struktur benötigen, um ihre biologischen Funktionen zu erfüllen.
Jedoch,Die Entdeckung intrinsisch ungeordneter Proteine (IDPs) und ihrer intrinsisch ungeordneten Regionen (IDRs)Dieses traditionelle Verständnis wird ständig neu gestaltet. Sie bilden unter physiologischen Bedingungen keine festen Strukturen, sondern sind tief in Kernprozesse wie die Signaltransduktion und die Regulation der Gentranskription involviert und stehen in engem Zusammenhang mit wichtigen menschlichen Krankheiten wie Krebs und neurodegenerativen Erkrankungen.
Untersuchungen der Computerbiologie haben zudem gezeigt, dass etwa 30% Aminosäurereste im eukaryotischen Proteom in einem ungeordneten Zustand vorliegen. Dies bedeutet, dass Unordnung nicht „abnormal“, sondern vielmehr ein normaler Bestandteil lebender Systeme ist.Aufgrund der hohen Dynamik ungeordneter Proteine ist es schwierig, diese mit traditionellen experimentellen Techniken stabil zu erfassen und ihre Konformationsverteilung mit herkömmlichen Rechenmethoden genau zu simulieren.Dies hat sich zu einem seit langem bestehenden technologischen Engpass in diesem Bereich entwickelt.
Um dieser Herausforderung zu begegnen, hat ein gemeinsames Team, bestehend aus Peptone, einem in Großbritannien ansässigen Entwickler von Proteinanalysetechnologien, der Universität Kopenhagen, NVIDIA, der Universität Oxford, dem MIT, der Duke University und anderen, zwei wichtige Durchbrüche vorgeschlagen.Eines davon ist das PeptoneBench-Systembewertungsmodell.Dieses Rahmenwerk integriert experimentelle Daten aus verschiedenen Quellen wie SAXS, NMR, RDC und PRE und kombiniert statistische Methoden wie die Maximum-Entropie-Neugewichtung, um einen strengen quantitativen Vergleich zwischen experimentellen Beobachtungen und theoretischen Vorhersagen zu erreichen.Das zweite ist das generative Modell PepTron.Das Training mit einem erweiterten synthetischen IDR-Datensatz verbessert insbesondere die Fähigkeit, ungeordnete Regionen zu modellieren, und ermöglicht so eine bessere Erfassung der Konformationsvielfalt ungeordneter Proteine.
Das Forschungsteam nutzte PeptoneBench, um PepTron systematisch mit gängigen Vorhersage-Tools wie AlphaFold2, Boltz2 und BioEmu zu vergleichen. Die Ergebnisse zeigten, dass PepTron eine hohe Übereinstimmung mit experimentellen Daten bei der Vorhersage geordneter und ungeordneter Bereiche aufwies und damit Bestleistungen erzielte. Basierend auf diesen Fortschritten entsteht ein präziseres und biologisch realistischeres Framework zur Vorhersage von Proteinstrukturen mithilfe eines „Konformationssatzes“, das unser Verständnis von Proteinen über das gesamte Spektrum von geordneten und ungeordneten Strukturen hinweg deutlich verbessert.
Die zugehörigen Forschungsergebnisse mit dem Titel „Advancing Protein Ensemble Predictions Across the Order–Disorder Continuum“ wurden als Preprint auf bioRxiv veröffentlicht.

Papieradresse:
https://www.biorxiv.org/content/10.1101/2025.10.18.680935v1
Folgen Sie unserem offiziellen WeChat-Account und antworten Sie im Hintergrund mit „PepTron“, um das vollständige PDF zu erhalten.
Weitere Artikel zu den Grenzen der KI:
https://hyper.ai/papers
Systematische Erstellung von PeptoneBench- und Multi-Source-Experimentaldatensätzen
Proteindatenbanken (PDBs) sind die grundlegendsten und wichtigsten öffentlichen Ressourcen in der Strukturbiologie, aber es gibt erhebliche strukturelle Lücken in ihrer Abdeckung von intrinsisch ungeordneten Proteinen (IDPs) und ihren ungeordneten Regionen (IDRs).Lediglich etwa 3%-Einträge wurden als ungeordnet markiert.Im menschlichen Proteom ist der Anteil solcher ungeordneter Regionen jedoch so hoch wie 20–30%.
Wie in der Abbildung unten dargestellt, führt diese systematische Verzerrung dazu, dass die meisten Strukturvorhersagemodelle stabile Konformationen naturgemäß „bevorzugen“, was ihre Fähigkeit, langfristig aus dynamischen, ungeordneten Zuständen zu lernen, einschränkt. Um diesen Mangel auszugleichen,Die Forscher führten ergänzende Datenbanken wie IDROMe ein, die einen unsortierten Anteil von etwa 771 TP3T enthalten.Sie kann die PDB hinsichtlich der statistischen Verteilung ergänzen. Allerdings fehlen dieser Datenbank Strukturdaten aus realen Experimenten, was ihre Verwendung als direkter Benchmark für Modellierung und Evaluierung erschwert und ihren Anwendungswert weiterhin deutlich einschränkt.
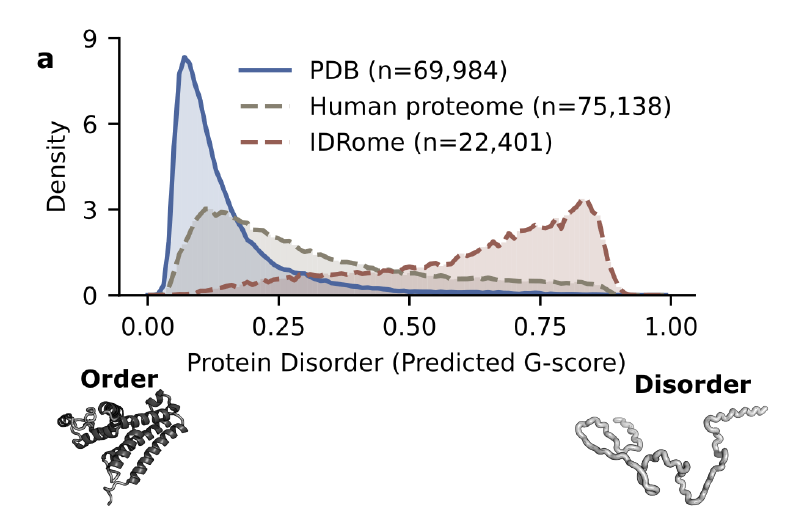
Um die zuvor genannten Datenengpässe zu überwindenDer erste Schritt besteht darin, quantifizierbare und vergleichbare Indikatoren für Unordnung zu ermitteln.Diese Studie verwendet den durchschnittlichen G-Score von Proteinen als zentrale Metrik. Die Werte reichen von 0 (vollständig geordnet) bis 1 (vollständig ungeordnet). Berechnet auf Basis von NMR-Daten zur sekundären chemischen Verschiebung (CS), spiegelt er präzise die Tendenz zur lokalen Sekundärstrukturbildung wider. Für Proteine, für die keine experimentellen CS-Daten vorlagen, nutzte das Forschungsteam das auf TriZOD trainierte maschinelle Lernmodell ADOPT2, um den G-Score vorherzusagen und so eine einheitliche Quantifizierung des gesamten Ordnung-Unordnung-Spektrums zu erreichen.
Darauf aufbauend wies das Team ferner darauf hin, dass die Qualität von Konformationssätzen nicht objektiv beurteilt werden kann, wenn man sich ausschließlich auf Strukturdaten aus PDBs stützt.Daher ist es notwendig, einen experimentellen Datensatz zu erstellen, der den gesamten Bereich von geordnet bis ungeordnet abdeckt.
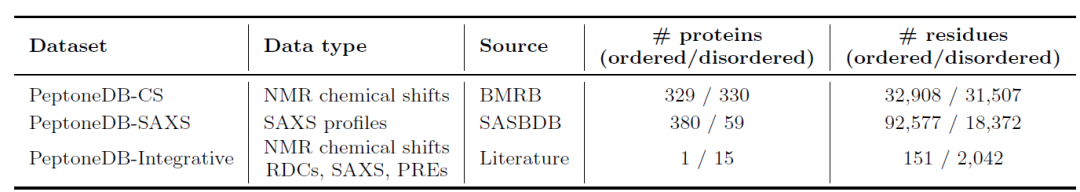
Zu diesem Zweck erstellten die Forscher, wie in der folgenden Tabelle dargestellt, drei komplementäre Datenquellen: PeptoneDB-CS (NMR-chemische Verschiebungen aus BMRB), PeptoneDB-SAXS (SAXS-Spektren aus SASBDB) und PeptoneDB-Integrative (ein spezieller IDP-Datensatz, der mehrere orthogonale experimentelle Daten integriert). Diese drei Datentypen weisen unterschiedliche Strukturen und komplementäre Informationen auf: CS zeigt lokale Strukturen, SAXS spiegelt die Gesamtkonformation wider und Integrative unterstützt die Kreuzvalidierung.
Basierend auf diesen Daten, wie in der folgenden Abbildung dargestellt.Forscher entwickelten das PeptoneBench-Evaluierungsframework, um die Übereinstimmung zwischen dem vorhergesagten Konformationssatz und experimentellen Daten zu quantifizieren.Der gesamte Prozess umfasst: Standardisierung und Vorverarbeitung der Konformationsdaten; Zuordnung der vorhergesagten Struktur zu experimentell vergleichbaren Beobachtungen mithilfe eines Vorwärtsmodells; Konsistenzbewertung basierend auf dem normalisierten RMSE unter Berücksichtigung der Unsicherheiten aus Modell und Experimenten. Die Endergebnisse werden als RMSE-G-Score-Diagramm dargestellt, und die Fehler werden mittels Lowes-Glättung und Bootstrapping geschätzt und zu einem PeptoneBench-Gesamtscore zusammengefasst. Dieser bildet einen quantitativen Standard für den direkten Vergleich der Leistungsfähigkeit verschiedener Tools.
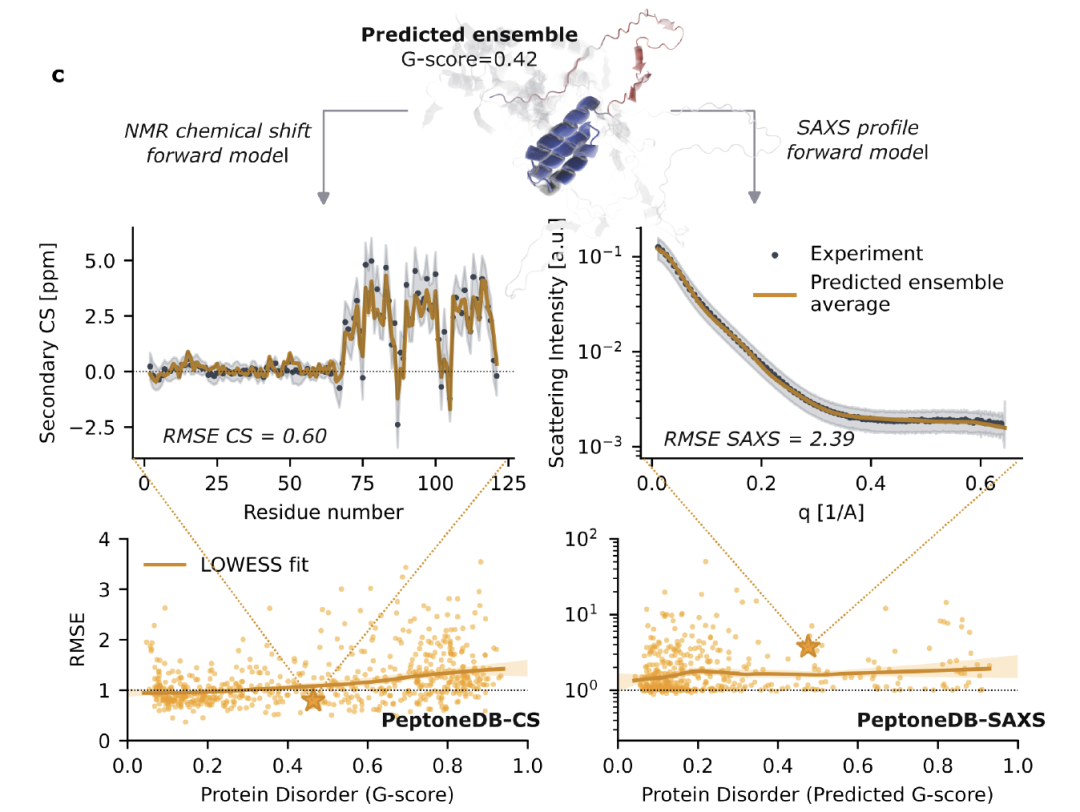
Es ist wichtig zu betonen, dass einige anfängliche Konformationssätze mit hohem RMSE nach einer Neugewichtung mittels maximaler Entropie tatsächlich näher an der experimentellen Verteilung liegen können. Um zu vermeiden, dass „falsche Gewichtungen“ fälschlicherweise als „fehlende Konformationen“ interpretiert werden,PeptoneBench gibt außerdem den RMSE vor und nach der Neugewichtung an, um zwischen korrigierbarer Stichprobenverzerrung und nicht wiederherstellbarem Konformationsverlust zu unterscheiden.Diese Strategie ist besonders wichtig für IDPs, die hochdynamisch und extrem empfindlich gegenüber experimentellen Bedingungen sind: Solange das generative Modell einen ausreichend reichen Konformationsraum abdecken kann, kann es sich durch den Neugewichtungsprozess schnell anpassen, selbst wenn die experimentelle Umgebung unterschiedlich ist, wodurch die Praktikabilität und Zuverlässigkeit der Vorhersageergebnisse deutlich verbessert wird.
PepTron: Ein Konformationsmodell, das geordnete und ungeordnete Proteine ausgleicht
Das vorgeschlagene PepTron-Modell ist ein Proteinkonformationsgenerator, der auf der ESMFlow-Flow-Matching-Architektur basiert. Sein Ziel ist es, das gesamte Konformationsspektrum von vollständig geordnet bis hochgradig ungeordnet abzudecken und eine Reihe von Konformationen zu generieren, die sowohl physikalisch plausibel als auch strukturell vielfältig sind.
Im Hinblick auf die Modellarchitektur,PepTron basiert auf ESMFlow und ist im NVIDIA BioNeMo-Framework implementiert, um die Effizienz des Trainings und der Inferenz zu verbessern.Das Modell integriert den cuEquivariance-Dreiecksaufmerksamkeitsmechanismus und unterstützt Flow-Matching-Funktionalität durch das Modular Co-Design-Subpaket von BioNeMo. Der Trainingsprozess folgt den Best Practices von BioNeMo für verteilte Systeme und kombiniert mehrere parallele Strategien sowie Berechnungen mit gemischter Präzision, wodurch eine stabile und effiziente Skalierung in Multi-GPU-Umgebungen ermöglicht wird.
Hervorzuheben ist, dass PepTron in der Inferenzphase weder auf multiple Sequenzalignments (MSA) noch auf externe ESM-Gewichte angewiesen ist. Es kann mit nur einem einzigen Prüfpunkt einen vollständigen Satz von Konformationen generieren, was die Anwendungsschwelle erheblich vereinfacht.
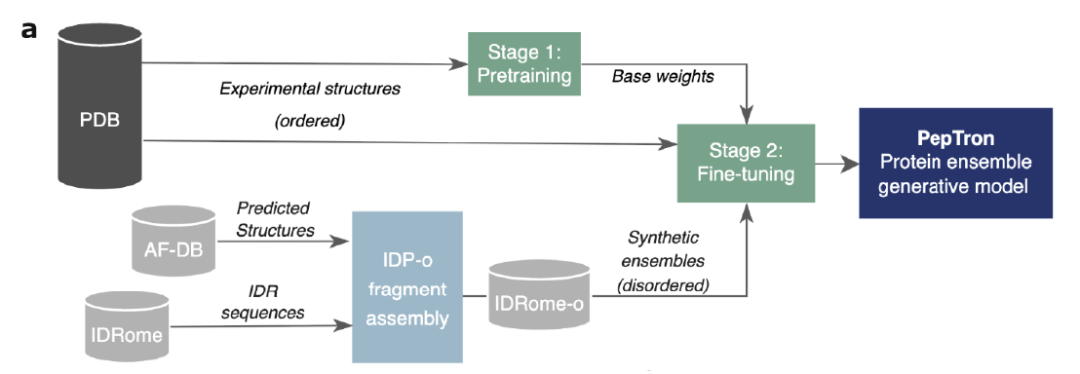
Um der Herausforderung der spärlichen experimentellen Strukturdaten in ungeordneten Bereichen zu begegnen, erstellte das Forschungsteam auf Basis von IDRome einen synthetischen Strukturdatensatz, IDRome-o. DaherSie entwickelten IDP-o, ein auf Fragmentassemblierung basierendes Werkzeug zur Generierung von Proteinstrukturen, das physikalisch plausible Sätze von IDP-Konformationen in großem Maßstab und zu extrem niedrigen Kosten erzeugen kann. IDP-o kombiniert Fragmentassemblierung und hierarchisches Kettenwachstum, um Sechs-Rest-Fragmente aus der AlphaFold-Datenbank zu extrahieren, die 214 Millionen Strukturen enthält, und erfasst so transiente helikale Strukturen in ungeordneten Proteinen genauer.
Es ist anzumerken, dass IDR-o nicht das Ziel verfolgt, eine bestimmte Gleichgewichtsverteilung zu simulieren, sondern alle möglichen Konformationen der Sequenz abzudecken. Daher eignet sich das Ergebnis besonders für die nachfolgende Maximum-Entropie-Neugewichtung und kann auch als hochwertige Ausgangskonformationsbibliothek für Molekulardynamiksimulationen dienen.
Um die Verzerrung traditioneller Modelle zu überwinden, die dazu neigen, stabile Strukturen vorherzusagen, wie in der folgenden Abbildung dargestellt,PepTron verwendet eine hybride Trainingsstrategie, die experimentelle und synthetische Daten kombiniert.Zunächst wird das Modell anhand experimentell ermittelter Strukturen aus der PDB-Datenbank vortrainiert. Anschließend wird ein synthetisch erzeugter Satz ungeordneter Proteine zur Feinabstimmung eingeführt, wodurch das Modell die kontinuierliche Verteilung geordneter und ungeordneter Konformationen vollständig erlernen kann. Selbst unter ressourcenbeschränkten Bedingungen verbessert diese Strategie die Vorhersageleistung des Modells für verschiedene Proteine signifikant.
Was konkrete Schulungsverfahren betrifft,Die Forschungsarbeit wurde in zwei Phasen unterteilt:In der ersten Phase wird, ausgehend von ESMFold-Gewichten, das Flow-Matching-Modul mithilfe von PDB-Daten neu trainiert und der Bereich für die Sequenzlängenkorrektur auf 512 Aminosäuren erweitert. In der anschließenden hybriden Feinabstimmungsphase dient ein hybrider Datensatz aus experimentellen PDB-Strukturen und synthetischen IDROMe-o-Daten als Trainingsdaten für die finale Optimierung des Modells. Dieses Design ermöglicht es PepTron, das gesamte Spektrum von geordneten und ungeordneten Strukturen abzudecken und so eine umfassendere und realistischere Modellierung des dynamischen Konformationsraums von Proteinen zu erreichen.
Modellvalidierung für Vollspektrum-Konformationen: Ein systematischer Vergleich von PepTron- und Mainstream-Methoden
Das Forschungsteam nutzte anschließend das PeptoneBench-Framework, um die Leistung von PepTron anhand experimenteller Daten systematisch und völlig unabhängig vom Trainingsdatensatz zu evaluieren und mit gängigen Modellen wie ESMFold, ESMFlow, AlphaFold2, Boltz2 und BioEmu zu vergleichen. Parallel dazu führte das Team spezifische Tests mit dem PeptoneDB-Integrative-Datensatz durch, der sich auf intrinsisch ungeordnete Proteine (IDPs) konzentriert, um die Fähigkeiten der einzelnen Modelle zur Modellierung ungeordneter Konformationen umfassend zu untersuchen. Die Ergebnisse zeigten deutliche Unterschiede zwischen den Modellen.
Wie in der folgenden Abbildung dargestellt, variiert die Leistung der einzelnen Modelle auf dem PeptoneDB-CS-Datensatz deutlich mit dem Grad der Proteinunordnung (G-Score): ESMFold und ESMFlow sind genau bei der Vorhersage geordneter Regionen, aber ihre Leistung ist in ungeordneten Regionen deutlich reduziert; IDP-o zeigt ein typisches komplementäres Muster - je höher der Grad der Unordnung, desto besser die Leistung.PepTron weist über das gesamte Konformationsspektrum von geordnet bis ungeordnet eine stabile, hohe Konsistenz auf.Diese Fähigkeit zum Ausgleich wurde im PeptoneDB-SAXS-Datensatz und der anschließenden neu gewichteten Analyse weiter bestätigt, was zeigt, dass PepTron die Konformationsvielfalt ungeordneter Proteine effektiv erfassen kann, ohne die Genauigkeit geordneter Strukturen zu beeinträchtigen.
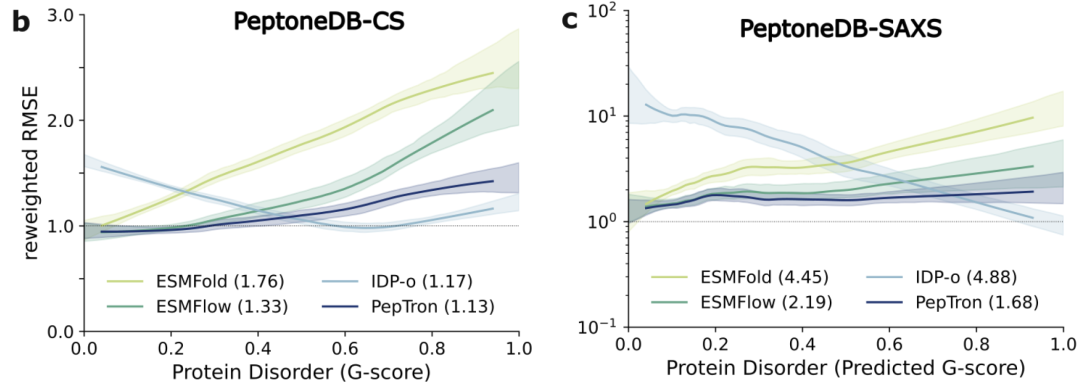
Weitere Ergebnisse des modellübergreifenden Vergleichs sind in der folgenden Abbildung dargestellt. Obwohl AlphaFold2 und Boltz2 bei der Vorhersage geordneter Proteine weiterhin führend sind, nimmt ihre Leistung systematisch mit zunehmendem Grad der Unordnung ab; im Gegensatz dazuPepTron und BioEmu weisen eine höhere Robustheit über das gesamte Konformationsspektrum auf und eignen sich daher besser für die Handhabung der hochgradig heterogenen Strukturmerkmale von IDPs.
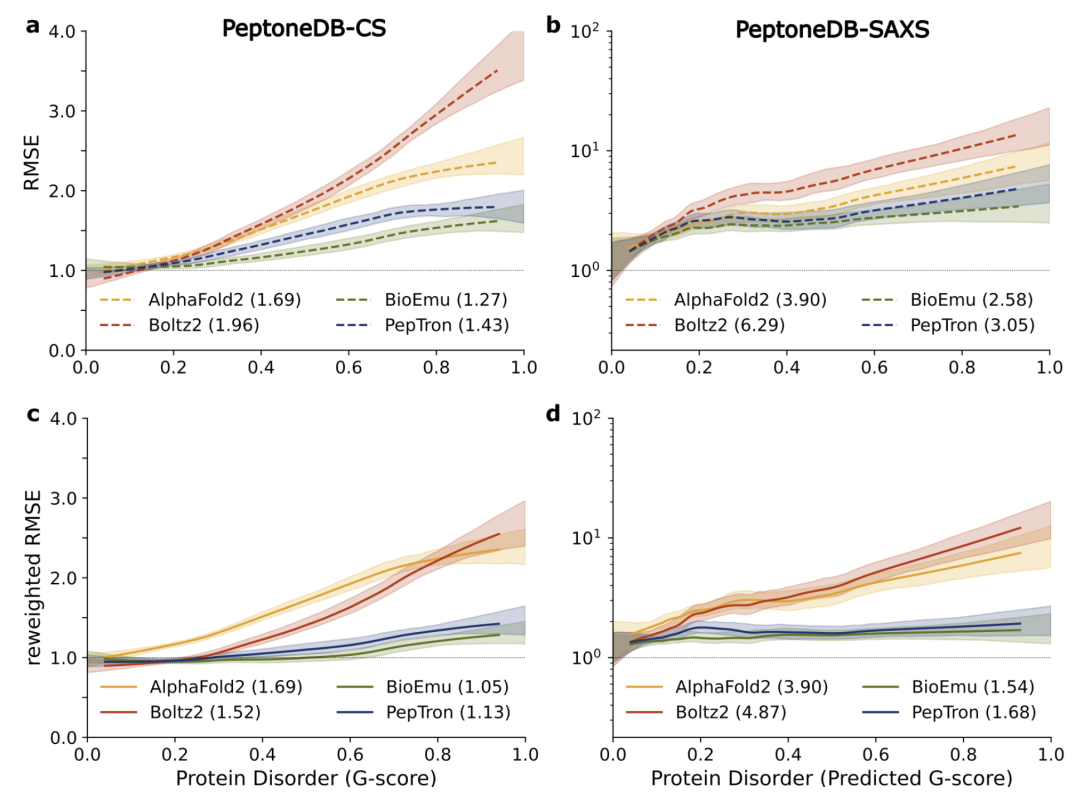
Um sicherzustellen, dass das Training an ungeordneten Regionen die Vorhersagekraft für geordnete Proteine nicht beeinträchtigte, führte das Forschungsteam zusätzliche Tests an geordneten Strukturdaten aus CAMEO22 und CASP14 durch. Die Ergebnisse zeigten, dass…PepTron schneidet bei wichtigen Kennzahlen wie RMSD, LDDT und TM ähnlich gut ab wie ESMFlow und beweist damit, dass es die Genauigkeit geordneter Strukturen nicht beeinträchtigt und gleichzeitig die IDR-Modellierungsfähigkeiten erweitert.
Im PeptoneDB-Integrative-Datensatz, der mehrere experimentelle Metriken integriert (siehe Abbildung unten), zeigen sich weitere Unterschiede in der Modellleistung. IDP-o schneidet nach der Gewichtung mit maximaler Entropie besonders gut ab und übertrifft andere Modelle sowohl im RMSE als auch im RDC-Q-Faktor deutlich. PepTron und BioEmu weisen ähnliche RDC-Metriken auf, BioEmu ist jedoch vorteilhafter bei der Vorhersage lokaler chemischer Verschiebungen. Es ist anzumerken, dass dies auch unter ungewichteten Bedingungen der Fall ist.IDP-o ist weiterhin führend bei den meisten lokalen und globalen Kennzahlen und beweist damit seinen natürlichen Vorteil bei der Abdeckung ungeordneter Proteinkonformationen.
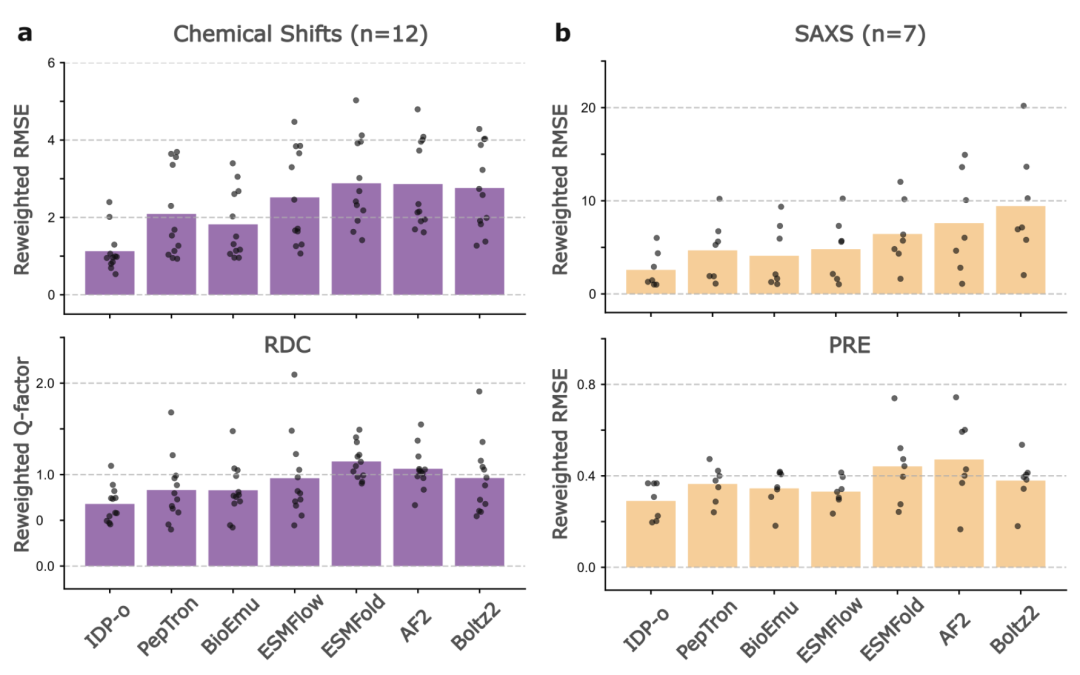
Die Studie wies zudem auf mehrere häufige Schwachstellen des aktuellen Modells hin:Die meisten Modelle erfassen Präferenzen für Fernkontakte nicht und weisen unterschiedliche Grade an Sekundärstrukturverzerrung auf. Darüber hinaus haben gängige Modelle im Allgemeinen Schwierigkeiten, den ungefalteten Zustand von „bedingt gefalteten Sequenzen“ präzise zu beschreiben, während IDP-o diesbezüglich eine einzigartige Überlegenheit aufweist.
Von Unordnung zu Ordnung: Globale Durchbrüche und neue Kapitel in der IDP-Forschung
Intrinsisch ungeordnete Proteine (IDPs) rücken aufgrund ihrer hochdynamischen Konformationseigenschaften und ihrer engen Verbindung mit vielen wichtigen Krankheiten immer mehr in den Fokus der Forschung in den globalen Lebenswissenschaften und der pharmazeutischen Industrie.
In der akademischen Forschung entwickelt sich die Technologie zur Vorhersage von KI-Strukturen zu einer Schlüsselrolle beim Knacken des „dynamischen Passworts“ von Identitätsdiebstählen.Die von der Universität Cambridge vorgeschlagene AlphaFold-Metainference-MethodeDurch die Kombination von AlphaFold-Ausrichtungsfehlerkarten mit Molekulardynamiksimulationen überwindet dieser Ansatz die Einschränkung des traditionellen AlphaFold, das hauptsächlich stabile Strukturen vorhersagt, und konstruiert erfolgreich IDPs und Sätze von Strukturen, die ungeordnete Bereiche enthalten, wodurch ein neuer Weg zum Verständnis ihres Polymorphismus eröffnet wird.
Titel des Artikels:
AlphaFold-Vorhersage von Struktur-Ensembles ungeordneter Proteine
Link zum Artikel:https://www.nature.com/articles/s41467-025-56572-9
Das Team der Universität Kopenhagen integrierte AlphaFold zudem in ein Protein-Sprachmodell.Es hat die großflächige Vorhersage der Konformation des ungeordneten menschlichen Proteoms ermöglicht.Dies beweist die Universalität und Skalierbarkeit der KI-Technologie in der IDP-Forschung.
Titel des Artikels:
Konformationsensembles des intrinsisch ungeordneten menschlichen Proteoms
Link zum Artikel:https://www.nature.com/articles/s41586-023-07004-5
Ob wissenschaftliche Erkenntnisse die Behandlung von Krankheiten tatsächlich verändern können, hängt von der Fähigkeit der Industrie ab, die Technologie in die Praxis umzusetzen. Die Zusammenarbeit zwischen dem britischen Biotechnologieunternehmen Peptone und dem deutschen Pharmaunternehmen Evotec…Dies zeigt einen gangbaren Weg auf, um die IDP-Forschung auf die Arzneimittelentwicklung auszuweiten.Mithilfe der ultraschnellen Wasserstoff-Deuterium-Austausch-Massenspektrometrie-Plattform (HDX-MS) von Peptone können Forscher die dynamischen Veränderungen ungeordneter Proteine in Echtzeit verfolgen und Bindungsstellen identifizieren, die mit herkömmlichen Strukturaufklärungsmethoden schwer zugänglich sind. In Kombination mit den Vorteilen von Evotec in den Bereichen Zielvalidierung, Wirkstoff-Screening und klinische Entwicklung ist es möglich, schwer zugängliche IDP-Zielmoleküle in potenzielle Wirkstoffkandidaten zu verwandeln.
Diese Fortschritte bestätigen nicht nur den Trend des PepTron-Modells, das das gesamte Spektrum geordneter und ungeordneter Strukturen abdeckt, sondern zeigen auch, dass ungeordnete Proteine, die einst als schwer fassbar galten, zunehmend zu wichtigen Zielstrukturen in der Präzisionsmedizin und der Biopharmazie werden. Dank kontinuierlicher technologischer Durchbrüche und einer vertieften Zusammenarbeit der Industrie könnten intrinsisch ungeordnete Proteine (IDPs) einen völlig neuen Rahmen für das Verständnis und die Entwicklung von Interventionsmöglichkeiten für zukünftige Krankheitsbehandlungen bieten.
Referenzlinks:
1.https://www.vbdata.cn/intelDetail/717834
2.https://c.m.163.com/news/a/JDIR2LQJ0552ZPM2.html
3.https://www.vbdata.cn/intelDetail/580634








