Command Palette
Search for a command to run...
Zur Unterstützung Der Proteinerzeugung/-faltung/-rückwärtsfaltung Schlugen Die Hunan University/University of Chinese Academy of Sciences/ByteDance Das APM-Modell Vor, Um Ein All-Atom-Design Und Eine Funktionsoptimierung Zu Erreichen
Als Hauptakteure des Lebens erfüllen Proteine ihre Funktionen oft in Form mehrkettiger Komplexe. Von der Antikörper-Antigen-Erkennung bis zur Enzym-Substrat-Bindung ist die präzise Interaktion zwischen mehrkettigen Proteinen entscheidend für das Verständnis der Lebensmechanismen.Allerdings weist das aktuelle Feld der KI-gesteuerten Proteinmodellierung eine erhebliche „Einzelketten-Verzerrung“ auf. Obwohl Modelle wie AlphaFold und die ESM-Reihe bahnbrechende Fortschritte bei der Faltung und dem Design von Einzelkettenproteinen erzielt haben, steckt die Modellierung von Mehrkettenkomplexen noch in den Kinderschuhen.
Bei bestehenden Methoden zur Verarbeitung mehrkettiger Proteine wird im Allgemeinen die Strategie der „Pseudosequenzverbindung“ angewendet, wodurch mehrkettige Proteine wie Einzelketten behandelt werden.Diese Methode schränkt die natürliche Ausprägung von Wechselwirkungen zwischen Ketten stark ein – in realen biologischen Komplexen lassen sich die Wechselwirkungen auf atomarer Ebene zwischen den räumlichen Positionen der Ketten und der Bindungsschnittstelle (wie Wasserstoffbrücken und hydrophobe Wechselwirkungen) nicht präzise durch lineare Verbindungen modellieren. Darüber hinaus ist die Generierung rein atomarer Strukturen mit zwei Herausforderungen verbunden: Die komplexe Konformation der Aminosäureseitenketten und die starke Sequenz-Struktur-Abhängigkeit erschweren die Neuentwicklung mehrkettiger Komplexe in diesem Bereich.
Um diese Forschungslücke zu schließen, haben die Universität Hunan, die Universität der Chinesischen Akademie der Wissenschaften und das ByteDance Seed-Team APM (All-Atom Protein Generative Model) vorgeschlagen, ein All-Atom-Proteingenerierungsmodell, das speziell für mehrkettige Proteinkomplexe entwickelt wurde. APM kann nicht nur direkt mehrkettige Komplexe mit rein atomaren Strukturen erzeugen, sondern unterstützt auch grundlegende Aufgaben wie Faltung und Rückfaltung und zeigt eine hervorragende Leistung beim Design funktioneller Proteine wie Antikörper und Peptide.
Die Forschungsergebnisse wurden unter dem Titel „An All-Atom Generative Model for Designing Protein Complexes“ für ICML 2025 ausgewählt.
Forschungshighlights:
* Native Modellierung mehrerer Ketten: Verzichten Sie auf Pseudosequenzverbindungen und lernen Sie direkt die Wechselwirkungen auf atomarer Ebene zwischen der unabhängigen räumlichen Verteilung mehrerer Ketten und der Bindungsschnittstelle.
* Optimierung der Darstellung aller Atome: Gleichen Sie Rechenleistung und Strukturdetails aus und erreichen Sie die Generierung einer Struktur auf atomarer Ebene durch die gemeinsame Darstellung von Aminosäuretyp, Rückgratgerüst und Seitenketten-Torsionswinkel.
* Verstärkung der Sequenz-Struktur-Abhängigkeit: Aufrechterhaltung der tiefen Verbindung zwischen Sequenz und Struktur durch Entkopplung des Rauschprozesses und bidirektionales Aufgabentraining (Falten/Entfalten).

Papieradresse:
Folgen Sie dem offiziellen Konto und antworten Sie mit „APM“, um das vollständige PDF zu erhaltenAPM-Datensatz zur Proteingenerierung:
Weitere Artikel zu den Grenzen der KI:
Datensatz: Umfangreiche Beispiele von einer einzelnen Kette bis zu mehreren Ketten
APM wird auf der Grundlage eines sorgfältig erstellten Proteindatensatzes aus mehreren Quellen trainiert, der die Struktur- und Sequenzinformationen von Einzelketten- und Mehrkettenproteinen integriert und so umfangreiches Lernmaterial für das Modell bereitstellt.
Der Single-Chain-Datensatz bietet eine umfassende Grundlage für die In-Chain-Modellierung durch Multi-Source-Fusion und Qualitätsfilterung. Er enthält insgesamt 187.494 Proben und deckt ein breites Spektrum an Proteintypen und Funktionskategorien ab. Die Daten stammen hauptsächlich aus drei maßgeblichen Datenbanken:
* PDB-Datenbank: Nach dem MultiFlow-Datenverarbeitungsprozess wurden 18.684 Proben gescreent;
* Swiss-Prot-Datenbank: hochwertige Strukturen mit pLDDT>85 ausgewählt und 140.769 Proben erhalten;
* AFDB-Datenbank: Unter Anwendung strengerer Screening-Kriterien wurden Proben mit pLDDT > 95 zurückbehalten, insgesamt 28.041 Proben.
Der Multi-Chain-Protein-Datensatz enthält insgesamt 11.620 Proben, die Proteinkomplexe mit 2–6 Ketten abdecken und wichtige Daten für die Multi-Chain-Modellierung liefern. Die Multi-Chain-Protein-Daten stammen aus PDB-Daten zu biologischen Assemblierungen (Biological Assemblies). Um Informationsverluste in nachgelagerten Prozessen zu vermeiden, schloss das Forschungsteam drei Arten von Proben aus: Proben, die in der SAbDab-Antikörperdatenbank vorhanden sind; Proben mit Kettenlängen unter 30 (als Peptide betrachtet); Proben mit einer Länge über 2.048 oder ohne Cluster-IDs.
Um die Generalisierungsfähigkeit des Modells zu verbessern, haben die Forscher die mehrkettigen Proben während des Trainingsprozesses zufällig gekürzt: Bei Proben mit mehr als 384 Resten wurden die nächsten 384 Aminosäuren beibehalten, zentriert auf den Restpaaren an der Bindungsschnittstelle zwischen den Ketten.Diese Bereinigungsstrategie stellt sicher, dass sich das Modell auf die wichtigsten Bindungsbereiche konzentrieren kann und gleichzeitig Speicherüberlaufprobleme vermieden werden.Darüber hinaus mischten die Forscher Einzelketten- und Mehrkettendaten proportional und nutzten die Fülle der Einzelkettendaten, um die Intraketten-Modellierungsmöglichkeiten zu verbessern. Jeder Probenentnahmeort ist mit umfangreichen Metadaten versehen, darunter geografischer Standort (Interketten-Interaktionsstelle), strukturelle Eigenschaften (wie Sekundärstrukturtyp) und Sequenzmerkmale (Aminosäuretyp und -konservierung). Diese Informationen liefern dem Modell mehrdimensionale Hinweise, um die Zuordnungsbeziehung zwischen Sequenz, Struktur und Funktion zu erlernen.
APM-Datensatz zur Proteingenerierung:
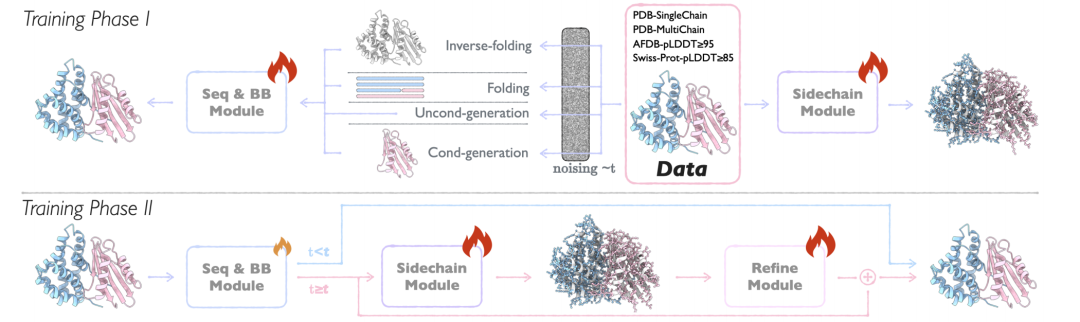
Modellarchitektur: ein kollaboratives All-Atom-Generierungsframework mit drei Modulen
Die Kernarchitektur von APM besteht aus drei Modulen mit klaren Funktionen: Sequence and Backbone Generation Module (Seq&BB Module), Sidechain Generation Module (Sidechain Module) und Refine Module.Durch innovatives Design wird eine End-to-End-Generierung von der Sequenz bis zur Allatomstruktur erreicht und gleichzeitig verschiedene Designaufgaben von Mehrkettenproteinen unterstützt.
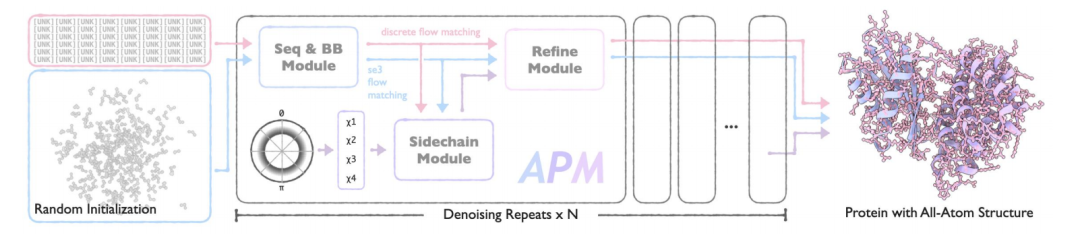
Seq&BB-Modul
Dieses Modul stellt die Grundlage von APM dar. Es verwendet die Flow-Matching-Methode, um die gemeinsame Generierung von Sequenz und Protein-Rückgrat zu realisieren und kann kollaborative Sequenz-Struktur-Modellierungsaufgaben auf Restebene bewältigen.Durch die Entkopplung des Rauschprozesses von Sequenz und Struktur wird die Beeinträchtigung der Sequenz-Struktur-Abhängigkeitsbeziehung reduziert. Die Faltungs-/Rückfaltungsaufgaben werden mit einer Wahrscheinlichkeit von 50% ausgeführt, um das bidirektionale Abhängigkeitslernen zu stärken. Die Kerninnovation des Moduls ist:
* Entkopplungsrauschen-Prozess:Durch die Trennung der Sequenz- und Strukturrauschprozesse wird die Zerstörung intermodaler Abhängigkeiten bei herkömmlichen Methoden vermieden. Die Rauschsequenz und das Rausch-Backbone werden unabhängig voneinander zu unterschiedlichen Zeitpunkten abgetastet. Dadurch wird sichergestellt, dass das Modell bidirektionale Sequenz-Struktur-Abhängigkeiten erlernen kann.
* SE(3) Flussanpassung:Im Hinblick auf die räumlichen Transformationseigenschaften des Proteinrückgrats wird eine dreidimensionale spezielle euklidische Gruppenflussanpassung (SE(3)) eingeführt, um die Translations- und Rotationsteile separat zu behandeln.
* Multitasking-Lernen:Es unterstützt außerdem bedingungslose Generierung, bedingte Generierung, Faltungs- und Rückfaltungsaufgaben und verbessert die Generalisierungsfähigkeit des Modells durch gemischtes Aufgabentraining. Die Verlustfunktion umfasst Flussanpassungsverlust und Konsistenzverlust, um die Glätte der generierten Flugbahn sicherzustellen.
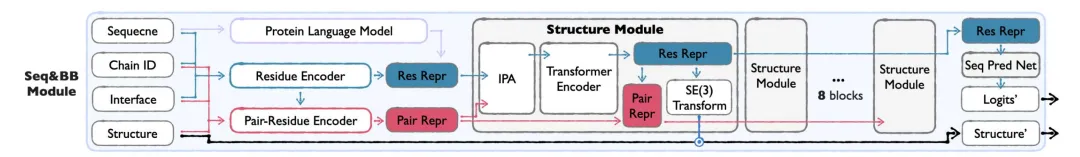
Sidechain-Modul
Um eine vollständige Atomstrukturgenerierung zu erreichen, sagt das Sidechain-Modul die Konformation der Aminosäureseitenketten basierend auf der von Seq&BB generierten Sequenz und dem Rückgrat voraus.
Das Modul verfolgt die folgenden Strategien:
* Torsionswinkel bedeutet:Die Seitenkettenstruktur wird durch Seitenketten-Torsionswinkel (bis zu 4 drehbare Bindungen) parametrisiert, wodurch Rechenleistung und Details auf atomarer Ebene in Einklang gebracht werden und die Komplexität der direkten Modellierung aller Atomkoordinaten vermieden wird.
* Zweistufige Ausbildung:Die erste Phase konzentriert sich auf die Aufgabe der Seitenkettenpackung und lernt die Verteilung der tatsächlichen Seitenkettenkonformationen; die zweite Phase wechselt zur Rekonstruktion tatsächlicher Seitenketten aus vorhergesagten Strukturen, um die Anwendbarkeit des Modells im Generierungsszenario sicherzustellen.
* Leichtbauweise:Im Vergleich zum Seq&BB-Modul verwendet das Sidechain-Modul weniger Strukturblöcke und kleinere versteckte Dimensionen.
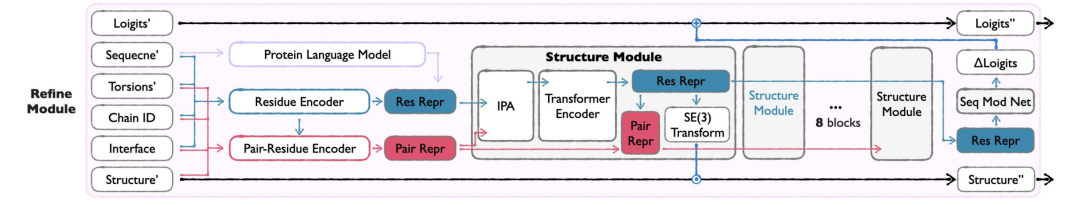
Modul „Verfeinern“
Als letztes Glied von APM integriert das Refine-Modul die Ausgabe von Seq&BB und Sidechain-Modul, optimiert die Sequenz und das Backbone durch Korrektur des Verlusts, reduziert atomare Konflikte und verbessert die strukturelle Rationalität.Die vollständigen Atominformationen werden verwendet, um die Sequenz und die Hauptkettenstruktur zu optimieren, Strukturkonflikte zu lösen und die generierten Ergebnisse dem natürlichen Protein anzunähern. Dieses Modul wird nur in der späten Generierungsphase (t≥0,8) aktiviert, um sicherzustellen, dass die Eingabequalität für die Optimierung ausreicht.
Experimentelle Schlussfolgerung: Mehrdimensionale Überprüfung der bahnbrechenden Leistung von APM
Die experimentelle Verifizierung von APM umfasst Single-Chain-Basisaufgaben, Multi-Chain-Kernaufgaben und nachgelagertes Funktionsdesign, und die Ergebnisse sind alle hervorragend.
Einzelkettenprotein-Aufgabe: Grundlegende Fähigkeiten vergleichbar mit professionellen Modellen
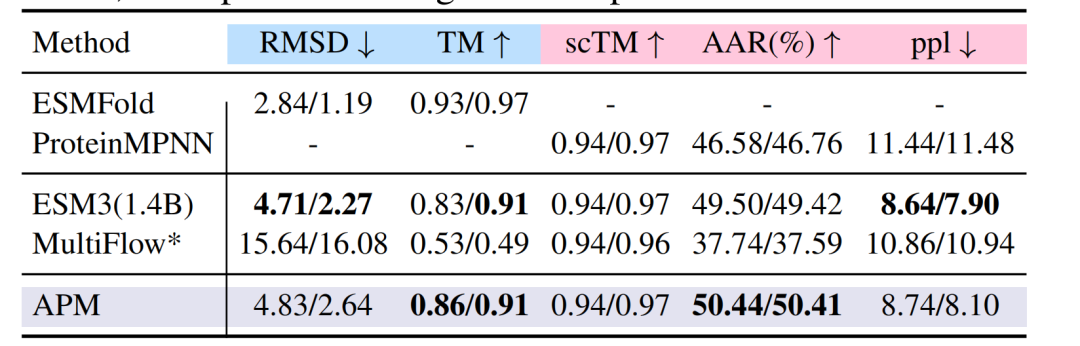
In der Faltungsaufgabe im PDB-DatensatzDer RMSD von APM beträgt 4,83/2,64,Der TM-Score erreichte 0,86/0,91, was mit der Leistung von ESM3, MultiFlow und anderen Modellen vergleichbar ist; bei der umgekehrten Faltungsaufgabe erreichte die Aminosäurerückgewinnungsrate (AAR) 50,44% und übertraf damit die 46,58% von ProteinMPNN.
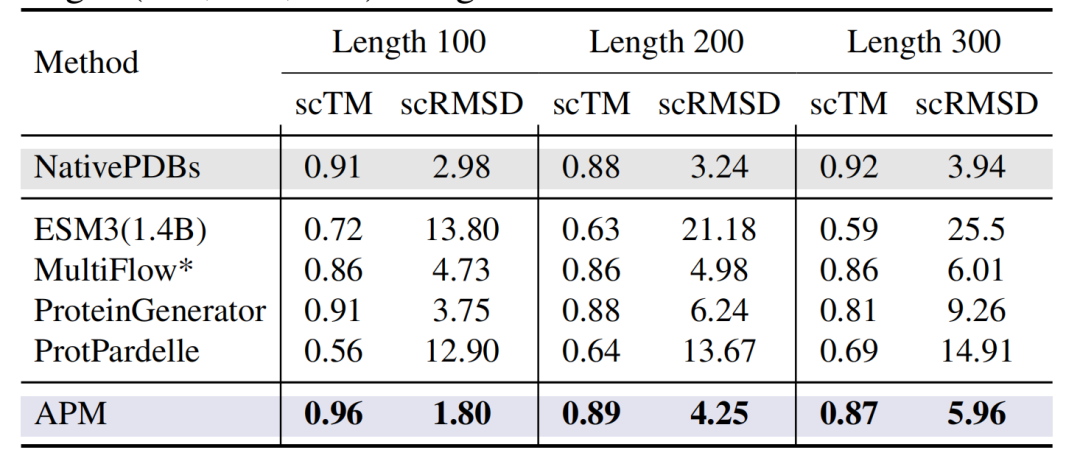
Darüber hinaus, wie in der Abbildung unten gezeigt, in den bedingungslos erzeugten Proteinen mit Längen von 100-300 Resten,Der scTM von APM beträgt bis zu 0,96 (Länge 100) und der scRMSD beträgt bis zu 1,80.Deutlich besser als All-Atom-Designmodelle wie ESM3 (1,4 B) und ProtPardelle.
Mehrkettenproteinaufgaben: Der Hauptvorteil der nativen Modellierung
In den Falt- und EntfaltungsexperimentenIm 2-6-Kettenkomplex beträgt die Faltungsleistung von APM 12,6/13,67 und ist damit niedriger als bei Boltz-1, übertrifft aber Boltz-1 ohne MSA deutlich. Die umgekehrte Faltung von scTM erreicht 0,85/0,95 und liegt damit nahe an Boltz-1 mit MSA. Dies belegt die Gültigkeit der Sequenz-Struktur-Assoziation. Die experimentellen Ergebnisse sind in der folgenden Abbildung dargestellt.
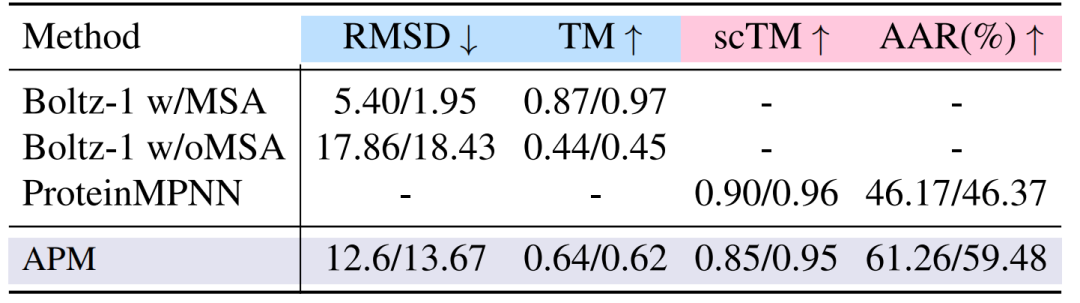
Zweitens,Der Mehrkettenkomplex hat eine starke Bindungsaffinität.Bei einer Kettenlänge von 50–100 erreicht die Bindungsenergie ΔG_RAA nach der Allatomrelaxation beispielsweise -112,65/-116,98 und ist damit deutlich besser als bei Chroma (-83,96/-86,66) und APM_BB (-114,94/-114,45), wenn nur die Hauptkette verwendet wird. Dies beweist die Notwendigkeit von Allatominformationen für die Modellierung von Wechselwirkungen zwischen Ketten.
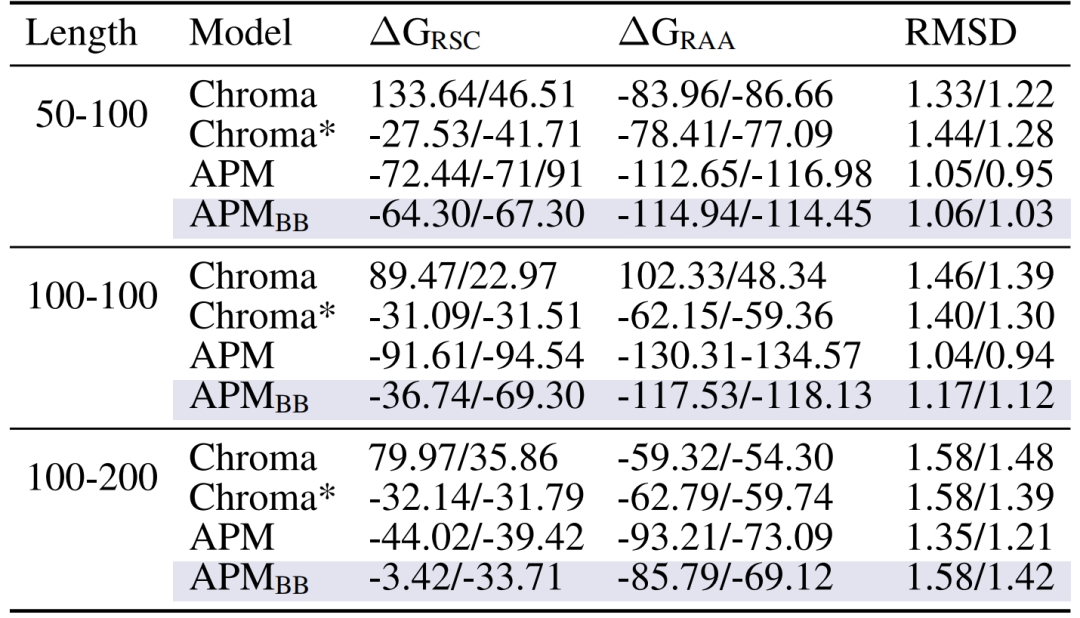
Interketten-Bindungsaffinität zwischen den erzeugten Komplexen
Downstream-Funktionsdesign: Durchbrüche bei der Anwendung von Antikörpern und Peptiden
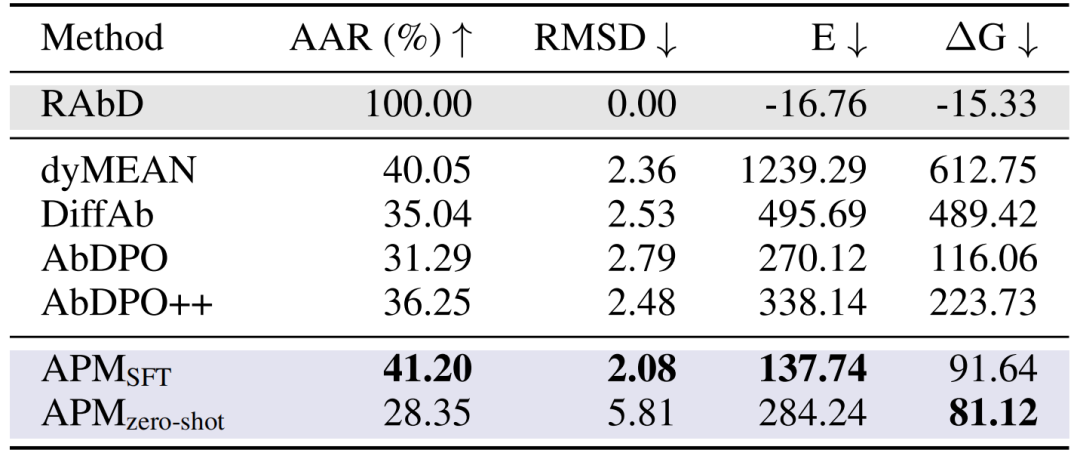
Antikörper CDR-H3-Design:Im RAbD-Benchmark-Test erreichte APMs AAR 41,20%, der RMSD lag bei 2,08 und die Bindungsenergie ΔG bei 91,64 und übertraf damit Methoden wie dyMEAN und DiffAb. Obwohl sich die Sequenz des von der Nullprobe erzeugten Antikörpers stark von der natürlichen unterscheidet, ist die Bindungsenergie besser (ΔG 81,12), was seine universelle Bindungsfähigkeit beweist.
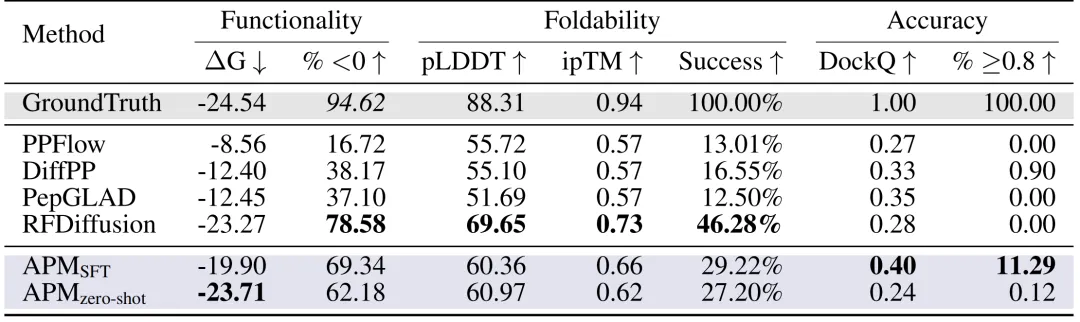
Peptiddesign:Anhand der PepBench- und LNR-Datensätze bewerteten die Forscher die Peptiddesignmethoden umfassend anhand von drei Schlüsselaspekten: Funktionalität, Faltbarkeit und Genauigkeit. Wie in der folgenden Abbildung dargestellt, erreichte die Bindungsenergie ΔG von APM (SFT) -19,90, 69,34%-Proben hatten ΔG < 0 und der Anteil von DockQ ≥ 0,8 erreichte 11,29% und übertraf damit PPFlow, PepGLAD und andere Methoden bei weitem. Die Faltungsstabilität (pLDDT 60,36, ipTM 0,66) war ausgezeichnet.
Zusammenarbeit zwischen Industrie und Forschung und Entwicklung führt zu Durchbrüchen in der Technologie zur Erzeugung von Proteinen auf Atombasis
Auf dem bahnbrechenden Gebiet der biologischen Proteinerzeugung aus reinen Atomen haben die Wissenschaft und die Wirtschaft nie aufgehört, es zu erforschen, und eine Reihe bahnbrechender Ergebnisse erregen weiterhin Aufmerksamkeit.
In der akademischen Welt hat AlphaFold3, das vom DeepMind-Team eingeführt wurde, durch die Integration mehrskaliger Strukturinformationen mit evolutionären Sequenzdaten starke Fähigkeiten auf dem Gebiet der All-Atom-Proteinerzeugung bewiesen.Präzise Modellierung komplexer Proteinfaltungsmuster,Insbesondere bei der Erzeugung von All-Atom-Komplexen mit Kofaktoren und Metallionen konnten Strukturgenauigkeit und Energierationalität im Vergleich zu herkömmlichen Methoden deutlich verbessert werden. Das vom Forschungsteam der Stanford University entwickelte ESM-IF1 verfolgt einen anderen Ansatz. Basierend auf einem impliziten Faltungsmodell, das mit umfangreichen evolutionären Sequenzdaten trainiert wurde, kann es direkt All-Atom-Proteinstrukturen mit natürlichen Konformationsmerkmalen erzeugen und eignet sich hervorragend für die präzise Konstruktion enzymatischer aktiver Zentren.
Auch die Wirtschaft engagiert sich in diesem Bereich und fördert industrielle Anwendungen durch technologische Innovationen. Die Beijing Bio-Geometry Biotechnology Co., Ltd. veröffentlichte das weltweit erste vollständige Proteinmodell auf atomarer Ebene – GeoFlow V2. Es ermöglicht die Entwicklung eines durchgängigen Diffusionsgenerierungs-Frameworks zur präzisen Regulierung von Proteinatomen. Im Vollatom-Design von Antikörper-CDR-RegionenEs kann Affinität und Stabilität gleichzeitig optimieren und so die Effizienz der Arzneimittelentwicklung deutlich verbessern.Das amerikanische Biotechnologieunternehmen Insilico Medicine hat ein Proteingenerierungssystem entwickelt, das sich auf die Entwicklung von Zielproteinen für Medikamente konzentriert. Die dabei verwendete Strategie zur Generierung mehrerer Einschränkungen ermöglicht die gezielte Optimierung der Bindungsstellen zwischen Proteinen und niedermolekularen Medikamenten und gewährleistet gleichzeitig die Rationalität der Atomstruktur. Dies bildet eine solide Grundlage für ein effizientes Screening von Medikamentenkandidaten.
Diese theoretischen Durchbrüche in der Wissenschaft und Anwendungsinnovationen in der Wirtschaft,Gemeinsam werden wir die Technologie zur Erzeugung von Proteinen auf Atombasis vom Labor in die industrielle Praxis überführen und so grundlegende Unterstützung für Durchbrüche in der präzisen Arzneimittelentwicklung, dem Design neuer Biokatalysatoren und der synthetischen Biologie leisten. Wir werden hoffentlich in Zukunft einen enormen Mehrwert für die Behandlung von Krankheiten und die Bioproduktion schaffen.
Referenzlinks:
1.https://mp.weixin.qq.com/s/a0bl9ek90t_-y8wy69Yu6Q
2.https://mp.weixin.qq.com/s/P-5o-R1qZY52Pq1yK5j6cQ








