Command Palette
Search for a command to run...
Das MIT-Team Macht BoltzGen Als Open Source Verfügbar Und Ermöglicht so Die Entwicklung Von Proteinbindern Für Verschiedene Molekültypen, Wobei Eine Nanomolare Affinität Für Das Ziel 66% Erreicht wird.

In den Bereichen Arzneimittelforschung und Biomolekulartechnik ist das De-novo-Binder-Design eine zentrale Methode für die automatisierte Arzneimittelforschung. Mithilfe von Computersimulation und Deep Learning können Forscher Peptid- oder Proteinstrukturen generieren, die an spezifische Ziele binden können. Dies ermöglicht die Entwicklung neuartiger Wirkstoffmodalitäten wie Antikörper, Nanobodies und zyklischer Peptide.
Traditionelle Proteindesign-Strategien basieren jedoch meist auf physikalischen Berechnungen wie molekulardynamischen Simulationen und Sequenzoptimierungsalgorithmen. Obwohl hohe Präzision in einem einzigen System erreicht werden kann,Allerdings ist der Rechenaufwand hoch, der Gestaltungsspielraum begrenzt und es ist schwierig, multimodale Ziele wie Proteine, kleine Moleküle und RNA gleichzeitig zu verarbeiten.Aktuelle tiefe generative Modelle haben zwar die Generierungsgeschwindigkeit bis zu einem gewissen Grad verbessert, verfügen jedoch im Allgemeinen nicht über die Fähigkeit zur strukturellen Schlussfolgerung auf atomarer Ebene und sind für bestimmte Molekülkategorien optimiert, was ihre Vielseitigkeit einschränkt. Darüber hinaus basiert die Modellbewertung häufig auf vorhandenen ähnlichen Komplexen im Trainingsset, was die Überprüfung der Generalisierungsfähigkeit für „unsichtbare Ziele“ erschwert. Ihnen fehlen kontrollierbare Generierungsmechanismen und flexible strukturelle Einschränkungsausdrücke, was zu Einschränkungen der Designeffizienz und Interpretierbarkeit führt.
Um dieses Problem zu beheben,Das MIT hat in Zusammenarbeit mit Boltz und anderen Institutionen das „All-atom Generative Model“ BoltzGen vorgeschlagen, das Strukturvorhersage und komplexes Design vereint.Dieses Modell ersetzt nicht nur herkömmliche diskrete Restbezeichnungen durch geometrische kontinuierliche Darstellungen, um ein gemeinsames Training der Proteinfaltung und des Bindungsdesigns in einem einzigen System zu erreichen, sondern konstruiert auch eine flexible „Design-Spezifikationssprache“, um eine kontrollierbare Generierung über Molekültypen hinweg zu erreichen.
Die experimentellen Ergebnisse zeigen, dassDie Nanobody- und Proteinkonjugat-Designs von BoltzGen zielen alle darauf ab, eine nanomolare Affinität für 66% zu erreichen.Zum ersten Mal wurde gezeigt, dass ein „Einzelmodellsystem“ eine gleichzeitige Optimierung der Faltungs- und Bindungsleistung beim multimodalen Biomoleküldesign erreichen kann.
Aktuell wurden die entsprechenden Forschungsergebnisse unter dem Titel „BoltzGen: Toward Universal Binder Design“ veröffentlicht.
GitHub-Adresse:
https://github.com/HannesStark/boltzgen
Forschungshighlights:
* Einheitliche Strukturvorhersage und Binderdesign in einem einzigen generativen All-Atom-Modell, das die gleichzeitige Proteinfaltung, Bindungsstellenmodellierung und Sequenzgenerierung mit atomarer Präzision ermöglicht und so die physikalische Rationalität und Steuerbarkeit des Moleküldesigns deutlich verbessert;
* Es wird eine universelle „Design-Spezifikationssprache“ vorgeschlagen, die es dem Modell ermöglicht, flexibel zwischen verschiedenen Systemen wie Proteinen, Nanoantikörpern, zyklischen Peptiden und kleinen Molekülen zu wechseln, die Generierung modalübergreifender Strukturen und die Kontrolle von Einschränkungen zu realisieren und den Anwendungsbereich der generativen KI im Bereich des biomolekularen Designs zu erweitern.

Papieradresse:
https://go.hyper.ai/3sx2K
Folgen Sie dem offiziellen Konto und antworten Sie mit „BoltzGen“, um das vollständige PDF zu erhalten
Weitere Artikel zu den Grenzen der KI:
Gemischte Datensätze: multimodale Trainingsstrategien
Das Forschungsteam verwendete beim Training von BoltzGen ein mehrstufiges, modalübergreifendes gemeinsames Trainingsframework.Die Kernquellen der verwendeten Datensätze umfassen drei Kategorien:
* Hochwertige experimentelle Strukturen aus der Protein Data Bank (PDB), die eine Vielzahl komplexer Strukturen wie RNA, DNA und kleine Proteinmoleküle abdecken und realistische chemische Bindungsbeschränkungen und dreidimensionale geometrische Verteilungsdaten für das Modell liefern;
* Experimentelle Daten aus der AlphaFold-Datenbank (AFDB), die von AlphaFold2 vorhergesagt und neu gelernt wurden und zuverlässige, durch Experimente generierte Faltmuster abdecken;
* Die vom Boltz-1-Modell generierten zusammengesetzten Strukturproben decken multimodale Szenarien wie die Bindung kleiner Moleküle und RNA-DNA-Interaktionen ab, was die Generalisierungsfähigkeit des Modells über verschiedene Biomolekültypen hinweg verbessern kann.
Um eine übermäßige Fokussierung des Modells auf bestimmte Strukturtypen zu verhindern, eliminierte das Forschungsteam hochskalierte Datensätze für Antikörper und TCRs, um die Diversität im generierten Raum zu erhalten. Darüber hinaus wurden alle Strukturproben während des Trainings zufällig ausgewählt und mehreren Aufgaben unterzogen, sodass das Modell in jeder Trainingsiteration Aufgaben wie Faltungsvorhersage, komplexes Design und Strukturvervollständigung zufällig bearbeiten konnte. Dieses einheitliche, multifunktionale Lernframework ermöglicht es dem Modell, Strukturen auf atomarer Ebene zu generieren und gleichzeitig über modalübergreifende Verständnisfähigkeiten zu verfügen.
Modellarchitektur: All-Atom-Inferenz vom Rauschen zur Struktur
Das Modell behält die Hauptkomponenten der AlphaFold3- und Boltz-2-Architekturen bei und nimmt auf dieser Grundlage einige Verbesserungen vor, um mehr bedingte Eingaben einzuführen.
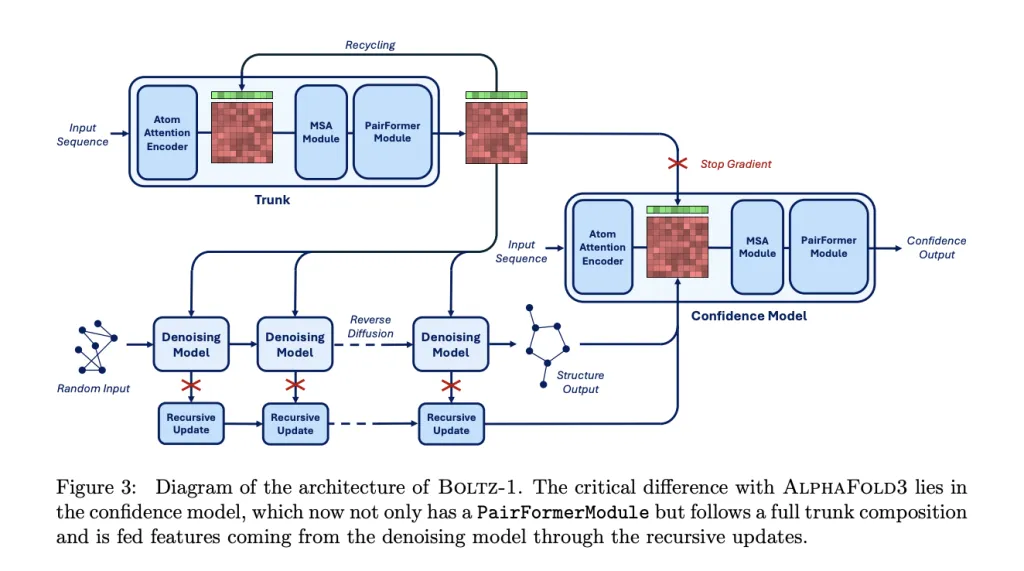
Wie in der folgenden Abbildung dargestellt, ist das gesamte Modell in zwei Hauptteile unterteilt:Ein größerer Trunk (Backbone-Netzwerk) und ein Diffusion Module (Diffusionsmodul).Das Trunk-Modul generiert Token- und paarweise Darstellungen zur bedingten Steuerung, während das Diffusionsmodul die 3D-Struktur basierend auf diesen Darstellungen generiert. Das Trunk-Modul wird nur einmal ausgeführt, während das Diffusionsmodul mehrere Iterationen durchführt, um die 3D-Koordinaten aller Atome schrittweise zu entrauschen.
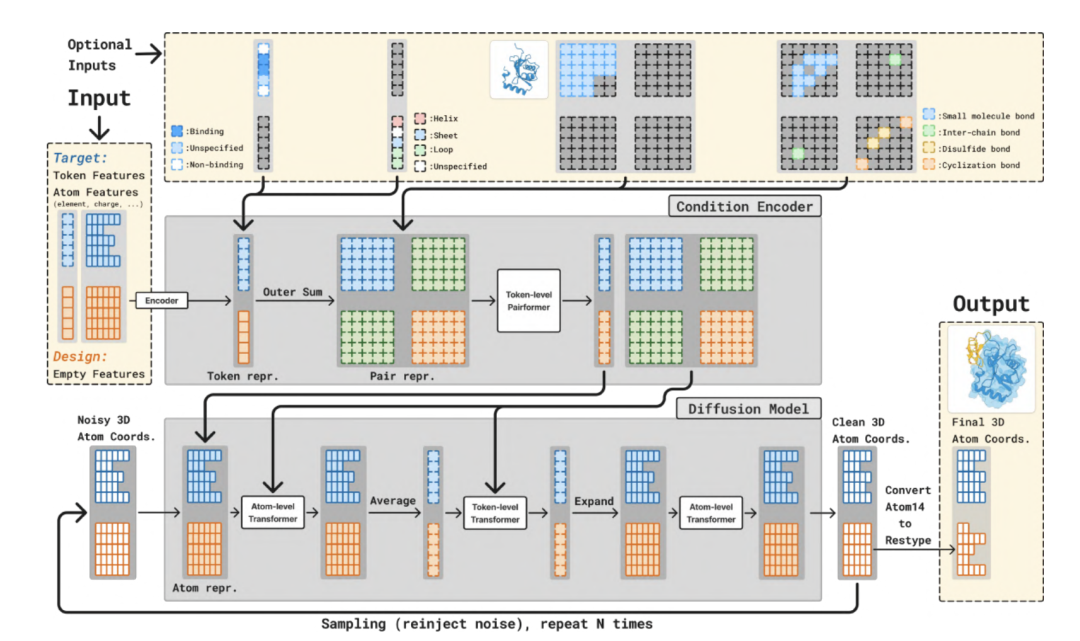
In der Trunk-Phase ähnelt es dem Trunk-Modul von Boltz-2 und ist für die Analyse der eingegebenen Proteinstruktur und Zielinformationen verantwortlich. Das Trunk-Modul verarbeitet tokenisierte Molekülstrukturen.Das Hauptframework verwendet eine PairFormer-Architektur und nutzt Triangle Attention, um die räumlichen Beziehungen zwischen Atomen effizient zu modellieren. In Kombination mit Geometric Residue Encoding werden gleichzeitig Resttypen und Atomkoordinaten in einem kontinuierlichen Raum abgeleitet, wodurch die Abhängigkeit von diskreten Aminosäuremarkierungen entfällt. Dieser Mechanismus ermöglicht es dem Modell, die physikalischen Gesetze der Struktur zum Zeitpunkt der Generierung wirklich zu verstehen, anstatt sich ausschließlich auf das Speichern von Daten zu verlassen.
In der Phase des DiffusionsmodulsDieses Modul empfängt verrauschte 3D-Atomkoordinaten als Eingabe.und prognostiziert dessen rauschfreie Koordinaten. Es verwendet eine Standard-Transformer-Architektur, die sowohl auf Atom- als auch auf Token-Ebene arbeitet. BoltzGen nutzt ein kontinuierliches Diffusionsmodell, um atomare Koordinaten schrittweise zu „entrauschen“. Es prognostiziert Rauschvektoren, um zufällige Anfangszustände in stabile Konformationen zu transformieren. Dabei bleiben die Beschränkungen der molekularen Energieoberfläche während des Generierungsprozesses erhalten, um physikalische Konflikte oder strukturelle Zusammenbrüche zu vermeiden.
Experimentelle Ergebnisse: Universelle Designvalidierung für 26 Ziele
Im experimentellen Teil deckte die Leistungsüberprüfung des BoltzGen-Modells mehrere Dimensionen ab, von Proteinen bis zu Peptiden, von neuen Krankheitserregern bis zu kleinen Molekülzielen, und demonstrierte eine hervorragende Generalisierung und Steuerbarkeit.
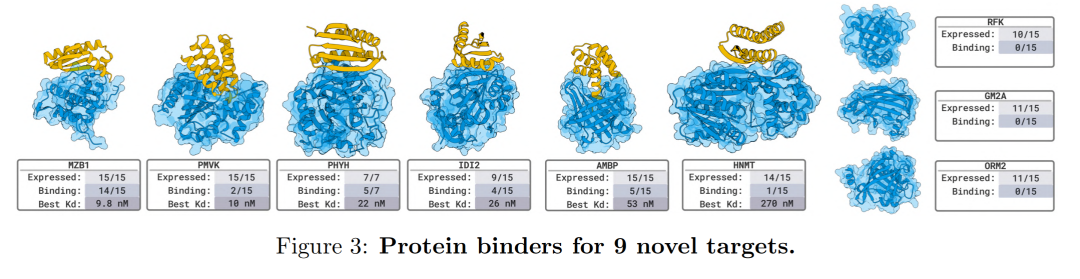
Das Team testete insgesamt 26 Ziele in 8 unabhängigen Wet-Lab-Validierungsprojekten.Die Ergebnisse umfassten eine Vielzahl von Bindungstypen, darunter Nanobodies, Proteine sowie lineare und zyklische Peptide. BoltzGen erzielte eine hohe Erfolgsquote bei unbekannten, komplexen Zielen: In neun Experimenten mit neuartigen Zielen, die sich völlig von den Trainingsdaten unterschieden, erreichten die entworfenen Proteine und Nanobodies alle eine nanomolare (nM) hochaffine Bindung an das 66%-Ziel. Dies demonstriert die leistungsstarken strukturellen Denk- und modalübergreifenden Designfähigkeiten des Modells.
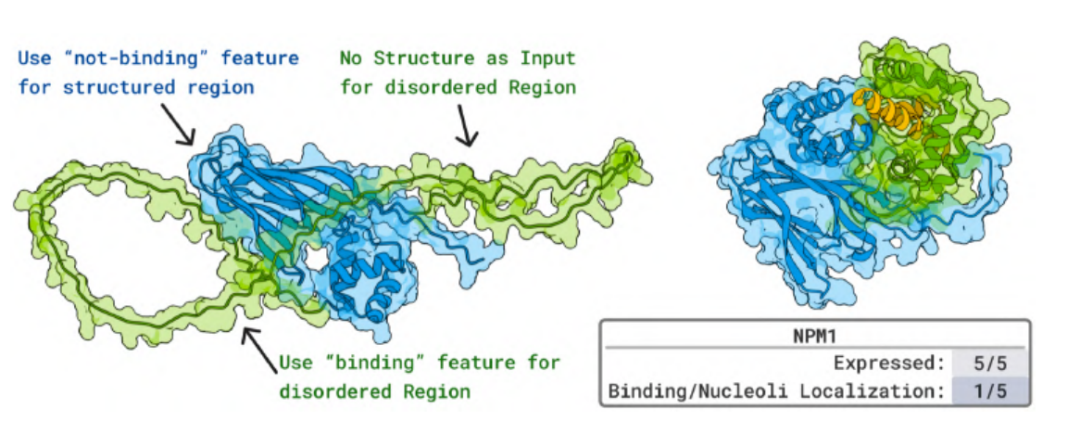
In Experimenten mit bioaktiven Peptiden unterschiedlicher Struktur,Von BoltzGen entwickelte Proteine können mit nanomolarer bis mikromolarer (μM) Affinität an verschiedene Arten von Peptidmolekülen binden und deren antimikrobielle oder hämolytische Aktivität effektiv neutralisieren. Für das ungeordnete Protein NPM1, das mit akuter myeloischer Leukämie assoziiert ist, zeigten die vom Modell generierten Peptide eine nukleoläre Kolokalisierung in lebenden Zellen. Dies liefert den ersten In-vivo-Beweis für die Fähigkeit von KI-entwickelten Proteinen, an natürlich ungeordnete Proteine zu binden.
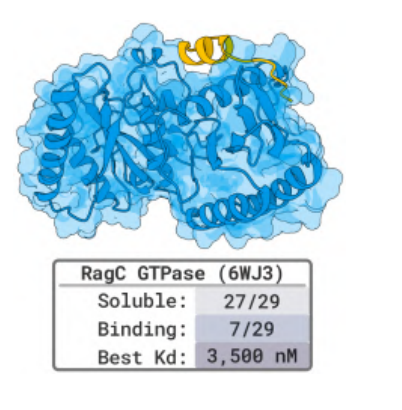
Auch das Design von RagC, einem zentralen Enzym des Zellstoffwechsels, und RagA:RagC-Dimeren führte zu bemerkenswerten Ergebnissen:Sieben der 29 Kandidatenpeptide banden erfolgreich an RagC, wobei die höchste Affinität 3,5 μM erreichte; 14 der Peptiddesigns mit zyklischer Disulfidbindung zeigten eine stabile Bindung.
BoltzGen demonstrierte außerdem maßstabsübergreifende Designfähigkeiten bei zwei kleinen Molekülen von biomedizinischem Interesse.Die resultierenden Proteinbinder zeigten eine nachweisbare Bindungsaktivität im Bereich von 50–150 µM und demonstrierten damit, dass das Modell eine Erkennung kleiner Moleküle ohne die Hilfe eines Chemieexperten erreichen kann. Darüber hinaus reduzierten Kandidatensequenzen über 19% bei der Entwicklung antimikrobieller Peptide, die auf die bakterielle DNA-Gyrase GyrA abzielen, das Bakterienwachstum um mehr als das Vierfache, wobei einige Peptide die Wirtszellen direkt abtöteten.
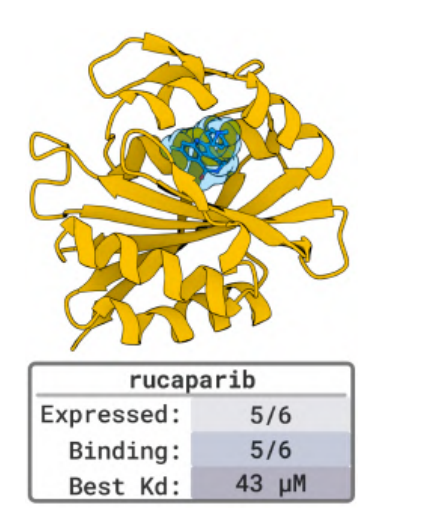
In den 5 Benchmark-Zieltests mit bekannten Bindungsstrukturen (wie PD-L1, TNFα, PDGFR usw.)BoltzGen erzielte außerdem eine hohe Trefferquote: Auf dem Ziel von 80% erschienen nanomolare Bindemittel, was bestätigt, dass seine Genauigkeit mit der des derzeit besten Modells vergleichbar ist.

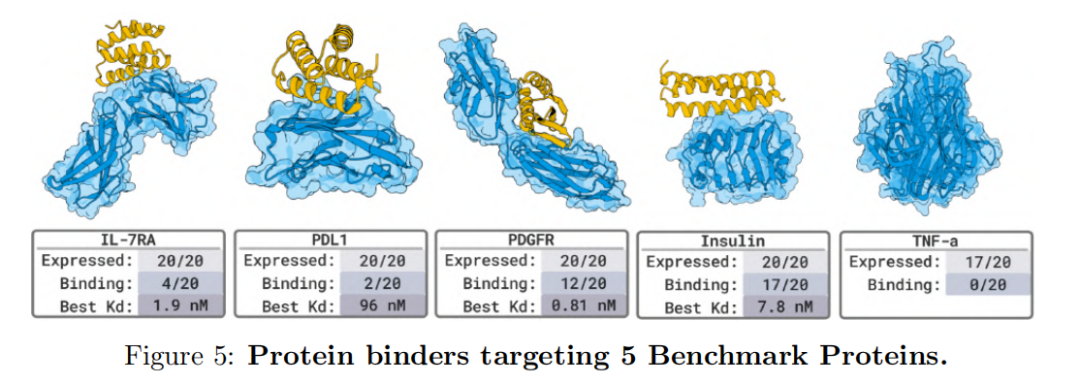
Insgesamt zeigt diese Versuchsreihe, dass BoltzGen nicht nur hochwertige Bindungsstrukturen innerhalb bekannter Datenverteilungen reproduzieren, sondern auch funktionales Design in völlig unbekannten biologischen Systemen erreichen kann. Seine einheitliche Architektur zur Generierung von Atomen integriert den Prozess „Design-Vorhersage-Verifizierung“ und bietet eine offene, kontrollierbare und skalierbare KI-Infrastruktur für die zukünftige Arzneimittelforschung und das Biomolekular-Engineering.
Von der Vorhersage bis zur Generierung verändert die Boltz-Reihe die Landschaft des KI-gesteuerten Moleküldesigns
Im Jahr 2024Das Forschungsteam der Jameel Clinic des MIT stellte das Boltz-1-Modell vor.Während sich die globale Arzneimittelentwicklungsbranche von der Strukturvorhersage zur Funktionsgenerierung verlagert und die AlphaFold-Modellreihe die Berechenbarkeit der Proteinfaltung vorangetrieben hat, schränkt die begrenzte Verfügbarkeit von AlphaFold3 die Möglichkeiten der Branche ein, in realen Arzneimittelszenarien frei zu iterieren. Boltz-1 wurde in diesem Kontext geboren. Es kommt nicht nur in der Leistung an AlphaFold3 heran, sondern ist auch vollständig Open Source und kommerziell rentabel, wodurch die Vorhersage molekularer Strukturen in das offene Ökosystem der Branche integriert wird.
Boltz-1 verwendet ein Generierungssystem, das ein Diffusionsmodell mit einer Transformer-Architektur kombiniert.Es kann die Strukturen von Proteinen, RNA, DNA und kleinen Molekülkomplexen auf atomarer Ebene vorhersagen. Seine flexible bedingte Schnittstelle ermöglicht die präzise Modellierung spezifischer Bindungsstellen oder Molekülkonformationen, was seine industrielle Anwendung erheblich erweitert. Von neuartigem Antikörperdesign und Enzym-Engineering-Optimierung bis hin zum Screening kleiner Molekülliganden können im Boltz-1-Framework End-to-End-Vorhersagen erzielt werden, was die Einstiegshürde für Biocomputing deutlich senkt.
Im Jahr 2025Das Team der Jameel Clinic des MIT stellte das auf Boltz-1 basierende Boltz-2-Modell vor.Es hat die Genauigkeit der Vorhersage der Proteinfaltung auf ein neues Niveau gehoben und ist als „GPT-4 der Strukturbiologie“ bekannt.
Im Vergleich zu seinem Vorgängermodell bietet Boltz-2 deutliche Verbesserungen bei der Generierungsgenauigkeit und Rechenleistung. Darüber hinaus ermöglicht es die multimodale bedingte Eingabe, die Sequenzinformationen, experimentelle Daten und chemische Eigenschaften integriert und so ein verfeinertes Moleküldesign ermöglicht. Da sich die globale Bioinformatik- und Arzneimittelforschungslandschaft in Richtung „vollständiger Szenariogenerierung“ entwickelt, trägt Boltz-2 der Nachfrage von Wissenschaft und Industrie nach hochverfügbaren, skalierbaren und kommerziell nutzbaren Werkzeugen Rechnung.
Boltz-2 übernimmt und optimiert das Hybriderzeugungssystem des Diffusionsmodells und der Transformer-Architektur.Sein Kernmodul Trunk kann mehrstufige Darstellungen von Protein- oder Nukleinsäurekomplexen gleichzeitig extrahieren.Das Diffusionsmodul generiert und optimiert darauf basierend die Struktur.
Dank einer flexiblen bedingten Schnittstelle können Forscher die Ausgabestruktur für spezifische Bindungsstellen, aktive Taschen oder kleine Molekülliganden präzise steuern. Dadurch erweitert sich das Anwendungspotenzial des Modells in Bereichen wie dem Design neuartiger Antikörper, der Optimierung der Enzymkatalyse und dem Screening von Wirkstoffkandidaten erheblich. Der Open-Source-Charakter von Boltz-2 gewährleistet zudem eine freie Iteration in Wissenschaft und Industrie und beschleunigt so die Anwendung molekularer generativer Berechnungen in realen Szenarien der Arzneimittelentwicklung.
Heute hat BoltzGen eine universelle „Design-Spezifikationssprache“ vorgeschlagen, die es dem Modell ermöglicht, flexibel zwischen verschiedenen Systemen wie Proteinen, Nanoantikörpern, zyklischen Peptiden, kleinen Molekülen usw. zu wechseln, um eine modalübergreifende Strukturgenerierung und Einschränkungskontrolle zu erreichen und so den Anwendungsbereich der generativen KI im Bereich des biomolekularen Designs weiter zu erweitern.








