Command Palette
Search for a command to run...
Die Tschechische Akademie Der Wissenschaften Veröffentlichte Das DreaMS-Modell, Das 200 Millionen Molekulare Massenspektren Umfasst, Um Den Weltweit Größten Massenspektrometrie-Datensatz GeMS Zu Erstellen
Laut Statistik beträgt der derzeit vom Menschen erforschte natürliche chemische Raum kleiner Moleküle weniger als 10% der Gesamtmenge, während in nicht zielgerichteten Metabolomik-Experimenten mehr als 90% an Massenspektren aufgrund fehlender zuverlässiger Anmerkungen zu „Datenmüll“ geworden sind.
In diesem entscheidenden Kampf um die Entschlüsselung von Molekülen besteht die zentrale Herausforderung darin, die komplexe Sprache der Tandem-Massenspektrometrie (MS/MS) zu entschlüsseln. Als hochmodernes Werkzeug der modernen chemischen Analytik ermöglicht das Flüssigchromatographie-Tandem-Massenspektrometrie-System (LC-MS/MS) eine effiziente Trennung von Molekülen mittels Flüssigchromatographie und erzeugt anschließend mithilfe der kollisionsinduzierten Dissoziationstechnologie Massenspektren von Fragmentionen. Dieser Prozess ähnelt der Zerlegung eines Moleküls und der Analyse seines Fragmentpuzzles.
Allerdings weisen die vorhandenen Analysewerkzeuge erhebliche Einschränkungen bei der Erstellung eines vollständigen molekularen Bildes auf:Sogar der fortschrittliche SIRIUS-Algorithmus ist zu stark von einer begrenzten Spektralbibliothek und künstlichen Regeln abhängig.Bei unbekannten natürlichen Molekülen, die insgesamt mehr als 801 TP3T ausmachen, besteht oft das Dilemma, dass keine Bibliothek zur Überprüfung zur Verfügung steht. Eine 2023 in Nature Methods veröffentlichte Studie wies darauf hin, dass in der globalen Metabolomik-Datenbank nur 21 TP3T-MS/MS-Spektren erfolgreich annotiert wurden und die verbleibenden 981 TP3T wie Riffe in der Tiefsee waren, was den Fortschritt der Arzneimittelforschung und der Krankheitsdiagnoseforschung erheblich behinderte.
Um dieses Problem zu lösen, nutzte ein Forschungsteam des Instituts für Organische Chemie und Biochemie der Tschechischen Akademie der Wissenschaften die bahnbrechenden Ergebnisse der GPT-Reihe im Bereich der Sprache und entwickelte einen speziellen Übersetzer für Massenspektren. Die Forscher analysierten 700 Millionen MS/MS-Spektren aus dem Global Natural Products Social Molecular Network (GNPS), erstellten erfolgreich den größten Massenspektrometrie-Datensatz aller Zeiten, GeMS, und trainierten das Transformer-Modell DreaMS mit 116 Millionen Parametern. Dieses Modell ist so, als würde künstliche Intelligenz die „gebrochene Grammatik“ von Molekülen von Grund auf lernen. Durch die Vorhersage maskierter Spektralspitzen und der chromatographischen Retentionsreihenfolge entdeckte es erfolgreich verborgene Strukturmuster in unmarkierten Massenspektren:Der von ihm generierte 1.024-dimensionale Charakterisierungsvektor kann die strukturellen Ähnlichkeiten zwischen Molekülen genau wiedergeben und zeigt eine hohe Robustheit gegenüber Signalschwankungen unter verschiedenen Massenspektrometriebedingungen.
Untersuchungen zeigen, dassDas fein abgestimmte DreaMS eignet sich gut für eine Vielzahl von Annotationsaufgaben im Bereich der Massenspektrometrie.Dazu gehört die Vorhersage spektraler Ähnlichkeiten, molekularer Fingerabdrücke, chemischer Eigenschaften und des Vorhandenseins von Fluor, die alle traditionelle Algorithmen und neu entwickelte Modelle des maschinellen Lernens übertreffen.DreaMS hat 201 Millionen Spektren integriert, um ein supermolekulares Netzwerk aufzubauen, das Bakterien, Pflanzen und menschliche Metaboliten abdeckt.Es wurde eine „molekulare Enzyklopädie“ für die chemische Gemeinschaft erstellt, die in Echtzeit aktualisiert werden kann und äußerst wertvolle Ressourcen für die Forschung und Anwendung in verwandten Bereichen bietet.
Die entsprechenden Forschungsergebnisse wurden in der international renommierten Fachzeitschrift Nature Biotechnology unter dem Titel „Self-supervised learning of molecular representations from millions of tandem mass spectra using DreaMS“ veröffentlicht.

Papieradresse:
Weitere Artikel zu den Grenzen der KI:
Download-Adresse des GeMS-Datensatzes zur chemischen Massenspektrometrie:
https://go.hyper.ai/IC2yw
GeMS-Datensatz: 700 Millionen Spektren zum Aufbau einer Massenspektrendatenbank
Die zentrale Datengrundlage dieser Studie ist der GeMS-Datensatz, der umfassend aus dem MassIVE GNPS-Repository gewonnen wurde und dessen Umfang und Qualität im Bereich der Metabolomik bahnbrechend sind.
Download-Adresse des GeMS-Datensatzes zur chemischen Massenspektrometrie:
https://go.hyper.ai/IC2yw
Wie in der Abbildung unten gezeigt,Das Forschungsteam integrierte 250.000 LC-MS/MS-Versuchsdaten aus biologischen und ökologischen Bereichen, extrahierte daraus etwa 700 Millionen MS/MS-Spektren und teilte sie durch strenge Qualitätskontrollalgorithmen in drei Untergruppen auf: GeMS-A, GeMS-B und GeMS-C.GeMS-A sammelt hauptsächlich Spektren mit dem 97% Orbitrap-Massenspektrometer, das den höchsten Qualitätsstandard repräsentiert. GeMS-C nutzt 52% Orbitrap- und 41% QTOF-Spektren, wodurch der Datenumfang deutlich erweitert und gleichzeitig eine bestimmte Qualität gewährleistet wird. Dieses hierarchische Design gewährleistet nicht nur die Zuverlässigkeit hochpräziser Instrumentendaten, sondern deckt durch umfassendere Teilmengen auch ein breiteres Spektrum an Massenspektrometrie-Technologiequellen ab und gewährleistet so die Vielfalt der Datensätze.
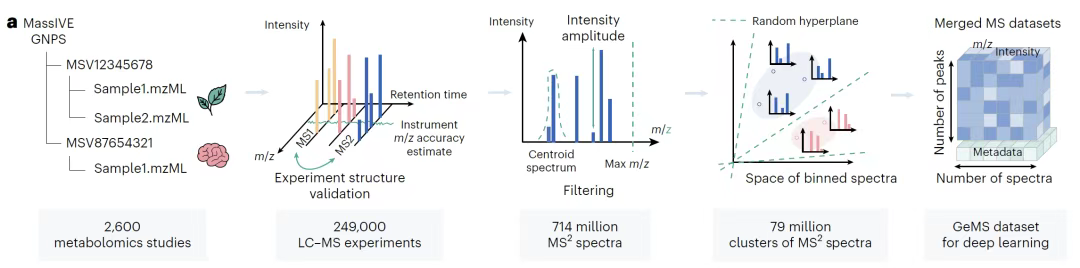
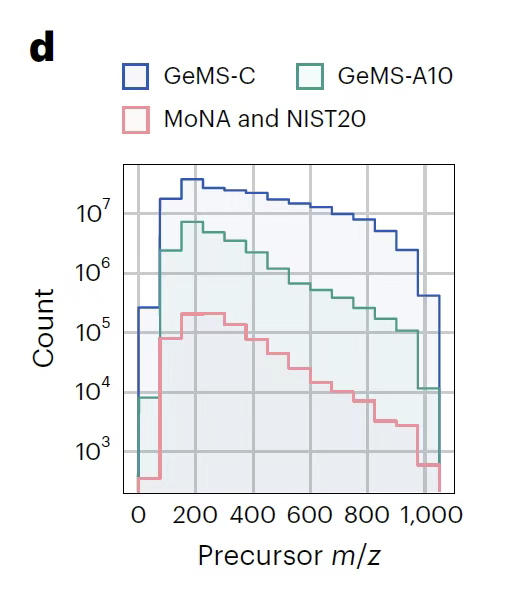
Um das Redundanzproblem bei großen Datenmengen zu lösen, nutzte das Forschungsteam den Locality Sensitive Hashing (LSH)-Algorithmus, um ähnliche Spektren effizient zu clustern. Durch Begrenzung der Spektrenanzahl im Cluster wurden neun Varianten generiert, wodurch die Rechenleistung optimiert und gleichzeitig die Repräsentativität der Daten gewahrt wurde. Der GeMS-Datensatz wurde schließlich im kompakten HDF5-Binärformat gespeichert.Konvertieren Sie das Rohspektrum in einen numerischen Tensor fester Dimension.Es überwindet den Größenengpass herkömmlicher Spektralbibliotheken – wie die Abbildung unten zeigt, ist sein Datenvolumen um mehrere Größenordnungen größer als das bestehender Bibliotheken, und seine Struktur ist hochgradig standardisiert, was beispielloses Trainingsmaterial für Deep-Learning-Modelle bietet. Diese Dateneigenschaften machen GeMS zum ersten ultragroßen Massenspektrometrie-Datensatz, der für unüberwachtes/selbstüberwachtes Lernen geeignet ist. Es legt nicht nur den Grundstein für das Vortraining des DreaMS-Modells, sondern bietet durch Qualitätsstratifizierung und Formatoptimierung auch präzise und umfassende Datenunterstützung für nachfolgende spektrale Ähnlichkeitsanalysen, die Charakterisierung molekularer Strukturen und andere Aufgaben. Dies fördert die Metabolomik-Forschung vom traditionellen Modell, das auf begrenzten Referenzbibliotheken basiert, zum intelligenten Analyseparadigma auf Basis massiver Rohspektren.
DreaMS-Modell: ein neues Paradigma für die Massenspektrometrieanalyse basierend auf einem selbstüberwachten Transformer
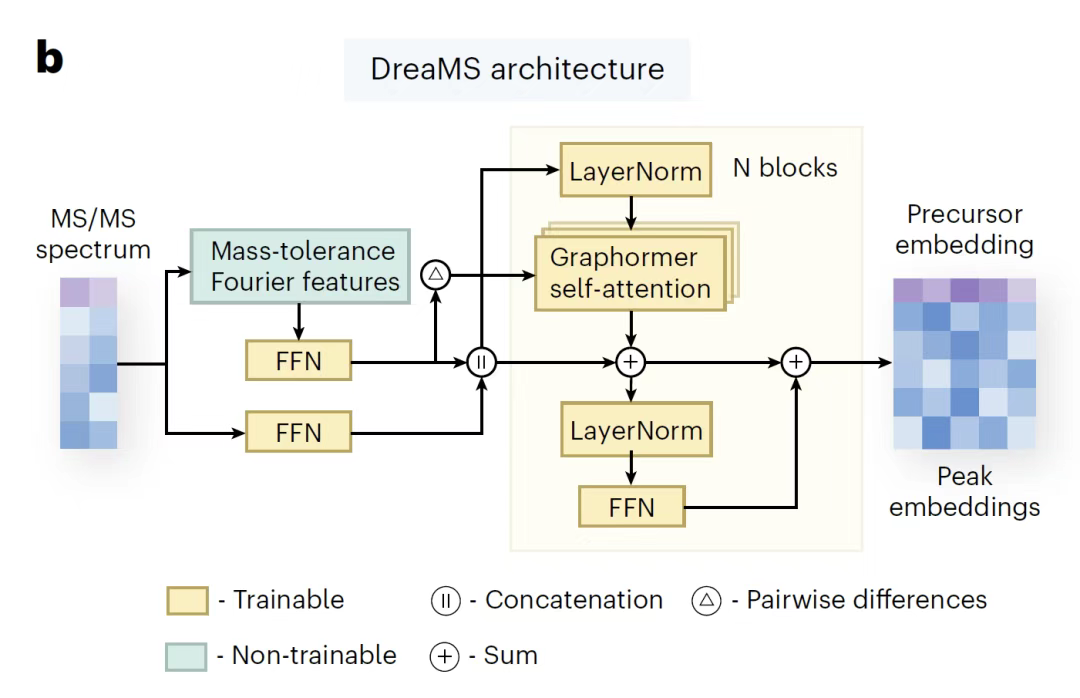
Basierend auf dem GeMS-Datensatz zielt das DreaMS-Modell darauf ab, durch selbstüberwachtes Lernen molekulare Darstellungen aus nicht annotierten MS/MS-Spektren zu extrahieren.Dieses Modell basiert auf der BERT-Architektur der natürlichen Sprachverarbeitung und war Vorreiter eines selbstüberwachten Lernparadigmas im Bereich der Massenspektrometrie kleiner Moleküle.Sein Kerndesign umfasst zwei Trainingsziele: Das eine besteht darin, das Masse-Ladungs-Verhältnis (m/z) von 30% im Spektrum im Verhältnis zur Intensität zufällig zu maskieren und das Modell zu trainieren, die maskierten Peaks zu rekonstruieren, während gleichzeitig „Elternionen-Tags“ eingeführt werden, um Informationen auf Spektralebene zu aggregieren (ähnlich der Darstellung auf Satzebene von Sprachmodellen); das andere besteht darin, zu lernen, die chromatographische Elutionsreihenfolge durch Spektralpaare desselben LC-MS/MS-Experiments vorherzusagen und die intrinsische Beziehung zwischen Molekülstruktur und Peakelutionsregeln zu stärken.
In Bezug auf die Modellarchitektur, wie in der folgenden Abbildung dargestellt,DreaMS basiert auf einem 7-Schicht-Transformer-Encoder, der mit einem 8-Kopf-Self-Attention-Mechanismus ausgestattet ist, der einen 1.024-dimensionalen Darstellungsvektor generieren kann.Für hochauflösende Masse-Ladungs-Verhältnisdaten nutzt das Modell die Vorverarbeitungstechnologie von Fourier-Merkmalen, um kontinuierliche Massenwerte in Sinus-/Cosinus-Frequenzkomponenten zu zerlegen, die Details von Ganzzahl- und Gleitkommaanteilen zu erfassen und die Elementzusammensetzungsvorhersagen über ein Feedforward-Netzwerk weiter zu verknüpfen. Der Intensitätswert wird von einem flachen Netzwerk verarbeitet und mit den Fourier-Merkmalen als Transformator-Eingang verknüpft. Darüber hinausDreaMS führt die Fourier-Merkmalsunterschiede aller Peakpaare explizit in den Self-Attention-Kopf ein (in Anlehnung an die Graphormer-Architektur).Modellieren Sie die neutrale Verlustbeziehung direkt und vermeiden Sie zusätzliche Beschriftungen oder komplexe Berechnungen.
In dieser Studie wurde eine lineare Sondierungstechnik verwendet, um Änderungen in den während der Trainingsphase erworbenen Darstellungen zu bewerten.Erstens kann das auf dem übergeordneten Ioneneinbettungsvektor basierende logistische Regressionsmodell während des Trainingsprozesses schrittweise den MACCS-Bindungsfingerabdruck vorhersagen, was darauf hindeutet, dass das Modell in Selbstüberwachung Informationen über Molekülfragmente lernt. Zweitens zeigt die Attention-Head-Analyse, dass das Modell den charakteristischen Peaks, die die Molekülstruktur darstellen, Vorrang vor dem Rauschen einräumt. Schließlich zeigen die Ergebnisse der Charakterisierungsraum-Clusterbildung, dass sogar die Spektren unter verschiedenen Ionisierungsbedingungen entsprechend der Molekülstruktur linear verteilt werden können, was seine Fähigkeit zur Erfassung struktureller Merkmale bestätigt.
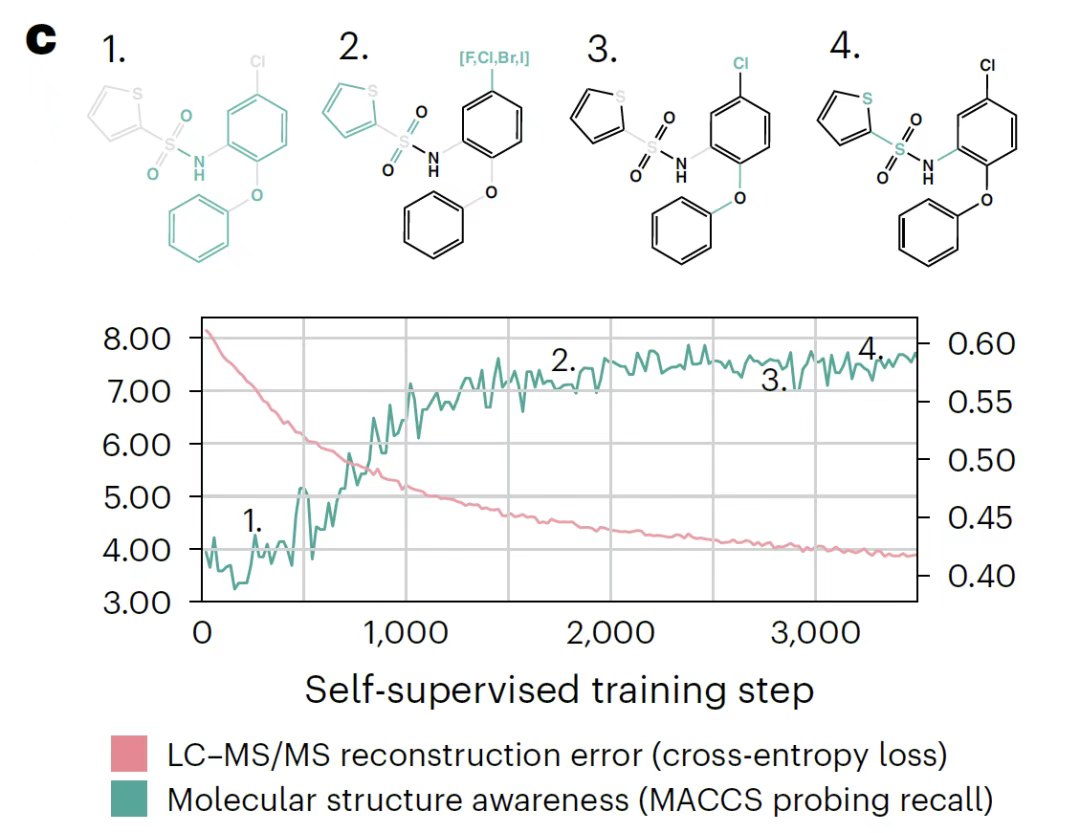
Aufgabenübergreifende Migration von DreaMS-Modellen: Massenspektrometrieanalyse von der Einzelmolekülanalyse bis zur vollständigen Metabolom-Verbindung
Als erstes Massenspektrometrie-Analysemodell, das auf selbstüberwachtem Lernen basiert, zeigte das DreaMS-Modell signifikante Vorteile bei der aufgabenübergreifenden Migration. Das Forschungsteam passte es an vier Kernaufgaben an:
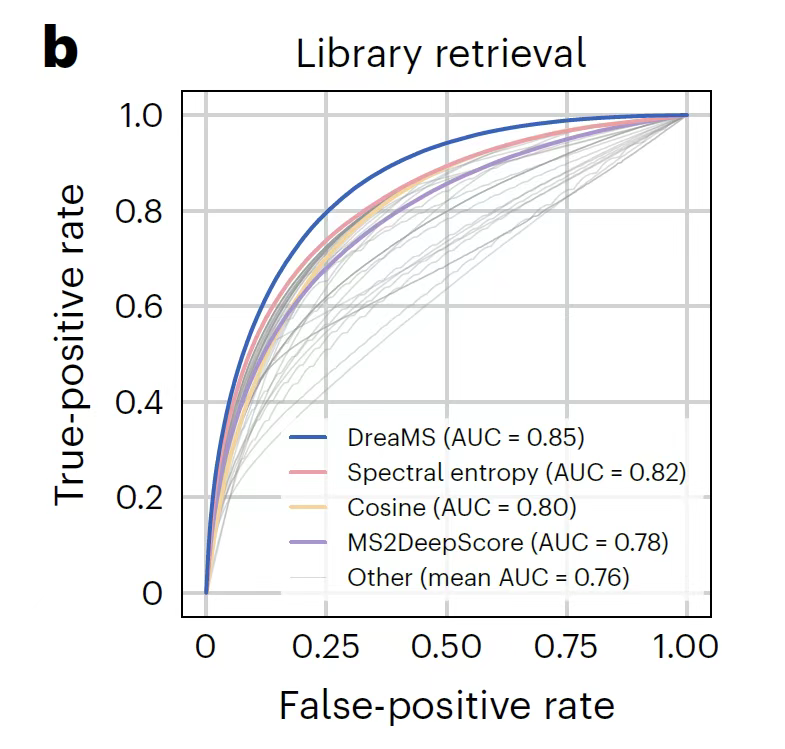
Bei der spektralen ÄhnlichkeitsanalyseWie in der folgenden Abbildung dargestellt, erreicht das Modell zunächst durch selbstüberwachte Charakterisierung eine Nullproben-Übereinstimmung. Die Korrelation zwischen der Kosinusähnlichkeit des Einbettungsraums und der Ähnlichkeit der Molekülstruktur (wie dem Tanimoto-Koeffizienten) übertrifft den überwachten Algorithmus MS2DeepScore, der auf dem Training mit markierten Daten basiert. Angesichts der Unempfindlichkeit von Nullproben gegenüber subtilen Unterschieden in der Molekülstruktur wird ein Triplett schwieriger Beispiele, darunter Referenzspektren, positive Proben desselben Moleküls und negative Proben ähnlicher Masse, zum Vergleich und zur Feinabstimmung entworfen, sodass bei der Abrufaufgabe mit einer Vorläufermassenabweichung innerhalb von 10 ppmDas fein abgestimmte DreaMS übertrifft 44 herkömmliche Ähnlichkeitsmetriken deutlich.Darüber hinaus sind die Einbettungsergebnisse robuster gegenüber Unterschieden bei Massenspektrometrie-Instrumenten und die UMAP-Analyse zeigt, dass ihr Darstellungsraum streng nach molekularen chemischen Formeln und Strukturmotiven gruppiert ist.
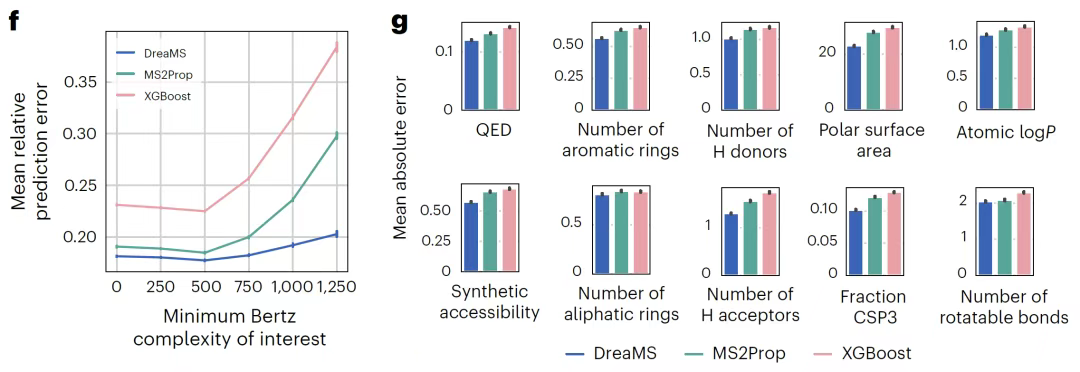
Bei der Aufgabe der Vorhersage molekularer FingerabdrückeWie die Abbildung unten zeigt, durchbricht DreaMS den komplexen Prozess herkömmlicher Methoden, die auf der Zuweisung chemischer Formeln oder der Generierung von Fragmentbäumen beruhen. Ein einziger Vorwärtsdurchlauf kann Morgan-Fingerabdrücke direkt aus Rohspektren vorhersagen. Die Suchleistung in der PubChem-Datenbank ist vergleichbar mit der des Deep-Learning-Modells MIST, das auf der Annotation chemischer Peak-Formeln basiert, jedoch auf rechenintensive Zwischenschritte verzichtet. Zur Vorhersage pharmazeutisch relevanter chemischer Eigenschaften gibt das Modell durch Feinabstimmung Lipinskis fünf Regelparameter, die molekulare Komplexität von Bertz und weitere Indikatoren aus.Es hat sowohl bei groß angelegten Arzneimittelscreenings als auch bei der Suche nach extraterrestrischen Biomarkern die derzeit beste Leistung erzielt.
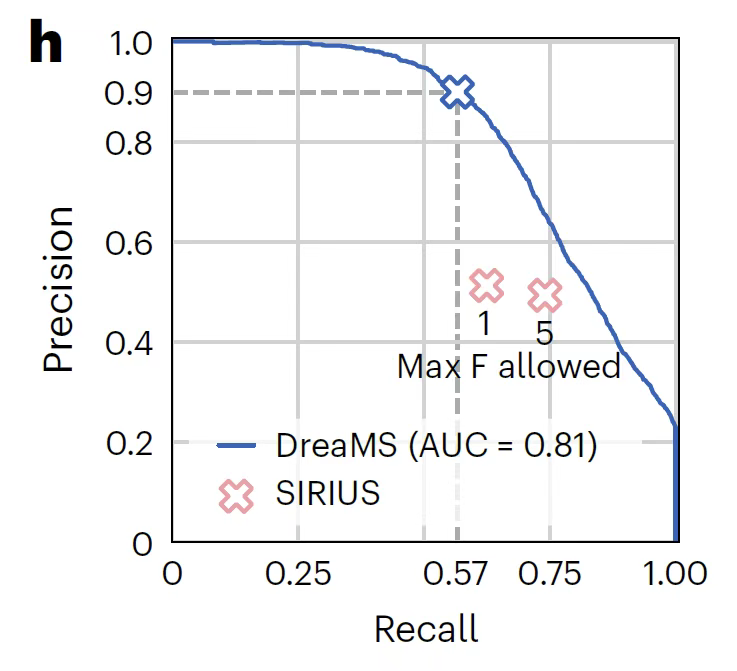
Bei der anspruchsvollsten Aufgabe, dem Nachweis fluorierter Moleküle,Wie in der folgenden Abbildung gezeigt, erreicht DreaMS durch ein probabilistisches Vorhersagemodell eine Präzision von 0,91 und einen Recall von 0,57.Dies ist dem SIRIUS-Algorithmus weit überlegen, der auf der Suche nach Fragmentierungsregelkombinationen basiert und eine Genauigkeit von nur 0,51 aufweist.Insbesondere weist es eine starke Generalisierungsfähigkeit bei der Erkennung von Molekülen mit neuartigen Strukturen auf und stellt ein wichtiges Instrument für die Entwicklung von Fluorid-bezogenen Arzneimitteln und die Umweltüberwachung dar.
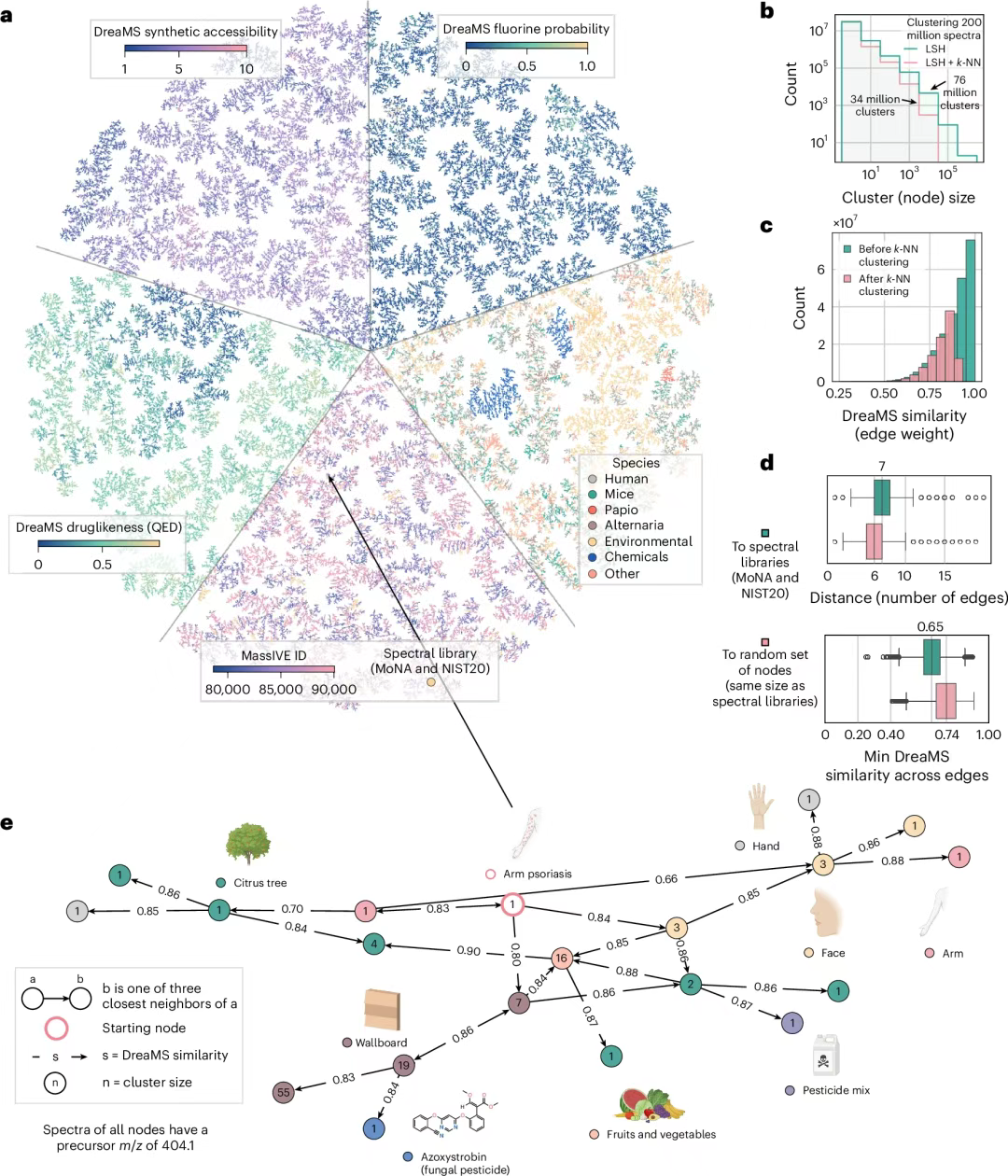
Basierend auf hoher Rechenleistung (die Einbettungsberechnung von 1 Million Spektren dauert auf der NVIDIA A100 GPU nur eine Stunde), wie in Abbildung ad unten dargestellt, erstellte das Forschungsteam einen DreaMS-Graph mit 201 Millionen Massenspektren und generierte durch lokales sensitives Hashing-Clustering einen 3-NN-Graph mit 34 Millionen Knoten. Die Kantenähnlichkeit von 67% liegt über 0,8, und 99,7%-Knoten bilden eine einzelne verbundene Komponente. Die kürzeste Pfadanalyse zeigt, dass jedes Spektrum innerhalb von sechs Schritten mit einem bekannten Bibliothekseintrag verbunden werden kann.
In einer Metabolomik-Studie zur Psoriasis am Arm,Wie in Abbildung e unten dargestellt, zeigt die Karte den möglichen Zusammenhang zwischen der Krankheit und dem Fungizid Pyraclostrobin durch spektrale Konnektivität. Der Assoziationspfad umfasst Umwelteinwirkungen wie kontaminierte Lebensmittel und behandelte Bäume und eröffnet eine neue datenbasierte Perspektive für die Erforschung der Ursachen komplexer Krankheiten. Die Fähigkeit, eine einzelne Aufgabe präzise zu beschreiben und so auf das gesamte Bibliotheksnetzwerk zu schließen, markiert eine neue Ära, in der sich die Massenspektrometrie-Analysetechnologie von der „Dekodierung einzelner Moleküle“ zur „Vernetzung des gesamten Metaboloms“ entwickelt hat.
Zusammenarbeit zwischen Industrie, Universitäten und Forschung treibt Innovationen in der Massenspektrometrie-Technologie voran
Im Bereich der Massenspektrometrieanalyse kleiner Moleküle und der Metabolomikforschung nutzen Universitäten und Unternehmen auf der ganzen Welt innovative Technologien, um Durchbrüche auf diesem Gebiet zu fördern.
Im Bereich der universitären Forschung gelang es mithilfe der KI-gestützten Multi-Omics-Big-Data-Analysetechnologie, die im Labor von Hu Zeping an der chinesischen Tsinghua-Universität entwickelt wurde, in Kombination mit hochpräzisen Metabolomik-Methoden, den metabolischen Interaktionsmechanismus zwischen Neuronen und Krebszellen im Tumormikroumfeld aufzudecken und Neurotransmitter-Regulationswege zu identifizieren, die als therapeutische Angriffspunkte genutzt werden können. Die Ergebnisse wurden mehrfach in Nature-Fachzeitschriften besprochen. Das „CataAI Characterization Expert System“, das vom Dalian Institute of Chemical Physics der Chinesischen Akademie der Wissenschaften entwickelt wurde,Durch die Integration von Deep-Learning-Technologie in den Prozess der Massenspektrometrie-Datenanalyse und die Verwendung selbst erstellter Datenbanken und neuer Algorithmen haben wir intelligente Empfehlungen von Massenspektren bis hin zu Molekülstrukturen erzielt.Für die komplexen Charakterisierungsdaten von Energiekatalysatormaterialien wurde ein zweistufiges neuronales Netzwerkmodell entwickelt.
Die Global Natural Products Social Molecular Network (GNPS)-Plattform der University of California, San Diego (UCSD)Als Quelle des Kerndatensatzes GeMS des in diesem Artikel untersuchten DreaMS-Modells fördert es weiterhin den einrichtungsübergreifenden Austausch und die Integration von Massenspektrometriedaten.Seine neueste Forschung etablierte eine Hochdurchsatzmethode zur Analyse der Metabolomik des Darmmikrobioms durch den Vergleich von Ethanol- und Methanol-Lösungsmittelsystemen und bietet so einen standardisierten Prozess zur Analyse der Interaktionsmechanismen zwischen Wirt und Mikrobe.
Im Rahmen der Unternehmensinnovationspraxis hat das amerikanische Unternehmen Agilent eine neue Generation von Systemen zur Flüssigkeitsqualitätserkennung wie die Pro iQ-Serie auf den Markt gebracht, die über hervorragende Leistung und Empfindlichkeit verfügen und sich ideal für die Überwachung komplexer biologischer Moleküle und die Erkennung von Verunreinigungen eignen.Sein Massenbereich wird auf m/z 2–3000 erweitert und seine Empfindlichkeit wird durch die Agilent Jet Stream (AJS)-Technologie verbessert.Es unterstützt die Routine- und Spurenerkennung kleiner Moleküle und Makromoleküle und bietet bahnbrechende technische Mittel zur Überwachung der Lebensmittelsicherheit. Basierend auf der Flüssigchromatographie-Tandem-Massenspektrometrie-Technologie hat das chinesische Unternehmen Kailaipu Technology unabhängig mehr als 20 klinische Massenspektrometrie-Kits entwickelt, die mehr als 300 Nachweiselemente abdecken, darunter die Nachweisreagenzien für Katecholaminmetaboliten in Blut und Urin, die in den Expertenkonsens der Chinese Medical Association Endocrinology Society aufgenommen wurden und zum klinischen Goldstandard geworden sind.
Generell erlebt die Massenspektrometrie kleiner Moleküle und die Metabolomikforschung derzeit einen technologischen Wandel, der von Universitäten und Unternehmen vorangetrieben wird. Diese Innovationen vertiefen nicht nur das Verständnis der Komplexität biologischer Systeme, sondern bieten auch großes Potenzial für praktische Anwendungen, von der Krebsfrüherkennung über die Prognose von Herz-Kreislauf-Erkrankungen und die Forschung und Entwicklung katalytischer Materialien bis hin zur Überwachung der Lebensmittelsicherheit. Diese durch die Verbindung von Algorithmeninnovation und experimenteller Wissenschaft ausgelöste Revolution könnte die gesamte Ökologiekette von der Grundlagenforschung bis zur klinischen Anwendung vollständig neu gestalten und damit weitreichende Auswirkungen auf verwandte Bereiche haben.
Abschließend möchte ich jedem eine Veranstaltung ans Herz legen: HyperAI veranstaltet am 5. Juli den 7. Meet AI Compiler Technology Salon in Peking.Wir haben das Glück, viele hochrangige Experten von AMD, der Peking-Universität, Muxi Integrated Circuit usw. eingeladen zu haben. Klicken Sie einfach auf den unten stehenden Link, um sich anzumelden.
https://www.huodongxing.com/event/1810501012111
Referenzartikel:
1.https://mp.weixin.qq.com/s/1QUjLMtj_6ui9T0gbuZtrA
2.https://dicp.cas.cn/xwdt/ttxw/202411/t20241107_7435521.html
3.https://ccms-ucsd.github.io/GNPSDocumentation/
4.https://mp.weixin.qq.com/s/Wgh2w0G76koqc9AY0PBHcg








