Command Palette
Search for a command to run...
Mit Weniger Als 100.000 Strukturierten Datenpunkten Trainierte Die Eidgenössische Technische Hochschule Lausanne (EPFL) Das PET-MAD-Verfahren, Das Eine Mit Professionellen Modellen Vergleichbare Genauigkeit Der Atomsimulation erreicht.
Von Halbleitermaterialien bis hin zu Wirkstoffmolekülen – die elektronische Struktur ist nach wie vor entscheidend für das Verständnis ihrer Eigenschaften. Ab-initio-Berechnungen auf Basis der Quantenmechanik ermöglichen die präzise Vorhersage von Struktur, Stabilität und Funktion von Materie und haben rasante Fortschritte im Materialdesign und in der Arzneimittelentwicklung vorangetrieben. Die Rechenkomplexität steigt jedoch mit der Systemgröße dramatisch an.Selbst hochmoderne Supercomputer haben Schwierigkeiten, komplexe Prozesse wie die Proteinfaltung und katalytische Reaktionen über lange Zeiträume zu simulieren.Dadurch ist eine Situation entstanden, in der „der Mechanismus zwar beschrieben, aber schwer zu berechnen ist“.
Da traditionelle Methoden an ihre Rechengrenzen stoßen, bietet maschinelles Lernen einen neuen Weg für Simulationen auf atomarer Ebene. Maschinelles Lernen von interatomaren Potenzialen, trainiert anhand quantenmechanischer Daten, bildet komplexe elektronische Strukturen auf effiziente Vorhersagemodelle ab und reduziert so die Rechenkosten um mehrere Größenordnungen bei nahezu gleichbleibender Genauigkeit. In den letzten zwei Jahrzehnten hat es in Systemen wie Hochtemperaturlegierungen und Biomakromolekülen signifikante Vorteile gezeigt und die Schwächen empirischer Kraftfelder hinsichtlich unzureichender Genauigkeit sowie die prohibitiv hohen Kosten von Ab-initio-Methoden überwunden.Frühe Modelle waren zumeist für einzelne Systeme konzipiert, was die wiederholte Generierung von Trainingsdaten und die Neuanpassung von Parametern erforderte und somit die systemübergreifende Forschung erheblich erschwerte.
Daher hat sich die Generalisierung zu einer zentralen Richtung für die weitere Entwicklung entwickelt. In den letzten Jahren sind verschiedene allgemeine Modelle entstanden, die sich an unterschiedliche Elemente und chemische Umgebungen anpassen lassen.Aufgrund unzureichender Trainingsdaten und uneinheitlicher Bewertungsstandards bleibt die Gesamtleistung jedoch instabil.
Vor diesem Hintergrund liefert das von der Eidgenössischen Technischen Hochschule Lausanne (EPFL) vorgeschlagene PET-MAD-Modell neue Beispiele.Durch die Nutzung eines Datensatzes, der ein breites Spektrum atomarer Diversität abdeckt, und einer Netzwerkarchitektur, die auf dem Point Edge Transformer basiert, erreicht es eine mit PET-Bespoke-spezifischen Modellen vergleichbare Genauigkeit, benötigt aber weit weniger Trainingsbeispiele als herkömmliche Modelle.Dies liefert einen eindrucksvollen Beweis für die Weiterentwicklung der Atomsimulation hin zu größerer Effizienz und breiterer Anwendbarkeit.
Die entsprechenden Forschungsergebnisse mit dem Titel „PET-MAD als leichtes universelles interatomares Potential für die Modellierung fortschrittlicher Materialien“ wurden in Nature Communications veröffentlicht.

Papieradresse:
https://www.nature.com/articles/s41467-025-65662-7
Folgen Sie unserem offiziellen WeChat-Account und antworten Sie im Hintergrund mit „PET-MAD“, um das vollständige PDF zu erhalten.
Weitere Artikel zu den Grenzen der KI:
https://hyper.ai/papers
MAD-Datensatz: Enthält 85 Elemente und fast 100.000 Strukturen
Der MAD-Datensatz wurde erstellt, um eine hochwertige Datengrundlage für das Training interatomarer Potentiale (MLIPs) im allgemeinen maschinellen Lernen zu schaffen. Er zeichnet sich durch eine breite chemische Vielfalt, strikte Datenkonsistenz und gute Verwendbarkeit aus.Es enthält 85 Elemente mit Ordnungszahlen von 1 bis 86 (ohne Astat), insgesamt 95.595 Strukturen.Es wird systematisch in 8 Untergruppen unterteilt, basierend auf seinen chemischen und strukturellen Eigenschaften, die im Großen und Ganzen Volumenkristalle, Oberflächen, Cluster, zweidimensionale Materialien und molekulare Systeme umfassen.
Der Kern des Datensatzes stammt aus einer Teilmenge der Materials Cloud 3D-Datenbank, genauer gesagt aus der MC3D-Teilmenge.Es enthält 33.596 individuelle Kristallstrukturen. Darauf aufbauend wurde eine MC3D-Rassel-Teilmenge (30.044 Strukturen) durch Störung der Atomkoordinaten mit Gaußschem Rauschen generiert, und eine MC3D-Zufalls-Teilmenge (2.800 Strukturen) wurde durch zufällige Neuanordnung der Atomarten im Kristall erstellt. Beide sind speziell darauf ausgelegt, extreme oder Nichtgleichgewichts-Verzerrungskonfigurationen besser abzudecken.
Um die Simulationsanforderungen für Oberflächen und niedrigdimensionale Systeme zu erfüllen, umfasst der Datensatz die MC3D-Oberflächen-Teilmenge (5.589 niedrigindizierte Kristallflächenmodelle) und die MC3D-Cluster-Teilmenge (9.071 Nanocluster), die durch Phasenschnitt gewonnen wurden. Die MC2D-Teilmenge (2.676 zweidimensionale Kristalle) wurde aus der Materials Cloud 2D-Datenbank importiert. Für molekulare Systeme stehen zwei Teilmengen zur Verfügung: SHIFTML-molcrys (8.578 Molekülkristalle) und SHIFTML-molfrags (3.241 Molekülfragmente). Alle diese Datensätze stammen aus maßgeblichen Datenbanken, um die Zuverlässigkeit der Proben zu gewährleisten.
Bezüglich DatengenerierungsstrategienMAD kombiniert direkte Wiederverwendung mit struktureller Modifikation. Vier Teilmengen – MC3D, MC2D, SHIFTML-molcrys und SHIFTML-molfrags – nutzen direkt veröffentlichte Strukturen, berechnen Energien und Kräfte jedoch mit identischen DFT-Parametern neu, wodurch systematische Abweichungen zwischen verschiedenen Quellen praktisch eliminiert werden. Die übrigen Teilmengen erweitern durch physikalisch motivierte Modifikationen der Grundstruktur (z. B. Hinzufügen von Rauschen, Umordnen von Atomen und Schneiden von Oberflächen) die Vielfalt der Konfigurationen gezielt.
PET-MAD: Architektur und Leistungsmerkmale eines universellen interatomaren Potentialmodells
Das PET-MAD-Modell wird auf einer Architektur trainiert, die durch die Pareto-Front optimiert wurde.Die Architektur besteht aus zwei Message-Passing-Schichten, die jeweils mit zwei Transformer-Unterschichten ausgestattet sind, welche 256-dimensionale Token-Repräsentationen, einen 8-Kopf-Multi-Head-Attention-Mechanismus und schließlich die Ausgabe des Ergebnisses über eine vollständig verbundene Schicht mit 512 Neuronen verwenden.Um die Stabilität des Trainingsprozesses zu verbessern, wird vor dem eigentlichen Training ein lineares Modell angepasst und der Basisbeitrag der Energie, der mit der atomaren Zusammensetzung zusammenhängt, subtrahiert.
Das gesamte Training wurde im PyTorch-Framework mit dem Metatrain-Paket durchgeführt. Die Rechenaufgaben wurden auf acht NVIDIA H100 GPUs verteilt, mit einer Batchgröße von 24 Architekturen pro GPU. Insgesamt wurden 1500 Epochen trainiert, was etwa 40 Stunden dauerte. Der Optimierungsprozess nutzte den Adam-Optimierer mit einer anfänglichen Lernrate von 10⁻⁴, die alle 250 Epochen halbiert wurde. Die Verlustfunktion bestand aus dem mittleren quadratischen Fehler der Energie- und Kraftvorhersagen, wobei der Energieterm mit 0,1 gewichtet wurde.
Nach dem Training erreichte das Modell einen durchschnittlichen absoluten Fehler von 7,3 meV/Atom für die Energie und 43,2 meV/Å für die Kraft im Trainingsdatensatz sowie 14,7 meV/Atom für die Energie und 172,2 meV/Å für die Kraft im Validierungsdatensatz, was eine ausgezeichnete Anpassungsgenauigkeit belegt.
Damit das allgemeine Modell besser an spezifische chemische Systeme angepasst werden kann,PET-MAD führt die Low-Rank Adaptation (LoRA) Fine-Tuning-Technologie ein.Anders als bei der traditionellen Feinabstimmung aller Parameter, die leicht zu „katastrophalem Vergessen“ (d. h. dem Verlust vorhandenen Allgemeinwissens) führen kann, friert LoRA alle Gewichte des Basismodells ein, indem es lediglich zwei Paare trainierbarer Matrizen niedrigen Rangs in jedes Aufmerksamkeitsmodul einfügt und deren Einfluss über einen Skalierungsparameter anpasst. Dadurch wird das Ziel einer „präzisen Verbesserung ohne Verlust der Allgemeingültigkeit“ erreicht. In dieser Studie ist der Rang von LoRA auf 8 festgelegt und der Skalierungsparameter auf 0,5 gesetzt.
Experimente zeigen, dassIn Szenarien mit begrenzten Daten übertreffen mit LoRA feinabgestimmte Modelle durchweg die speziell für dieses System von Grund auf trainierten Modelle.Selbst in komplexen Systemen wie Bariumtitanat können feinabgestimmte Modelle physikalische Observablen erzeugen, die mit dedizierten Modellen vergleichbar sind, und dabei die allgemeine Genauigkeit beibehalten. Daher werden sie als Optimierungsschema für spezifische Anwendungen empfohlen.
Für allgemeine Modelle ist eine zuverlässige Fehlerschätzung entscheidend. PET-MAD quantifiziert die Unsicherheit mithilfe der Last Layer Prediction Rigidity (LLPR)-Methode. Diese Methode schätzt den a posteriori-Fehler neuer Vorhersagen, indem sie die Kovarianz der letzten Schicht der verborgenen Merkmale des Modells auf dem Trainingsdatensatz analysiert und dabei nahezu keinen zusätzlichen Rechenaufwand verursacht. Noch wichtiger ist, dass LLPR die Auswahl einer endlichen Anzahl von Gewichten der letzten Schicht ermöglicht, um ein ressourcenschonendes „flaches Ensemble“ zu bilden. Dadurch wird die Unsicherheit auf die Berechnung komplexer abgeleiteter Größen wie der freien Energie und Phononendispersionskurven übertragen. Diese Methode ist deutlich recheneffizienter als herkömmliche Ensemble-Methoden und eignet sich daher hervorragend für die praktischen Anforderungen großskaliger Atomsimulationen.
Auch,PET-MAD bietet zwei Kraftvorhersagemodi: Der eine basiert auf automatischer Differenzierung (Backpropagation), um die Kraft aus der Energieanalyse abzuleiten, wobei die Energieerhaltung strikt eingehalten wird; der andere sagt die Kraft direkt aus den Atomkoordinaten mithilfe eines separaten neuronalen Netzwerkmoduls voraus, was die Inferenzgeschwindigkeit um das 2- bis 3-fache verbessern kann, jedoch eine geringe Nichterhaltung der Energie mit sich bringen kann.Durch die Entwicklung eines speziellen Mehrzeitschritt-Integrators konnte das Forschungsteam die durch den letztgenannten Modus verursachten Abtastfehler effektiv vermeiden und so sowohl die Recheneffizienz als auch die Zuverlässigkeit der dynamischen Simulation gewährleisten.
Sowohl Benchmark- als auch praktische Vorteile: Erreichen der Genauigkeit eines dedizierten Modells mit minimalem Bedarf an proprietären Daten.
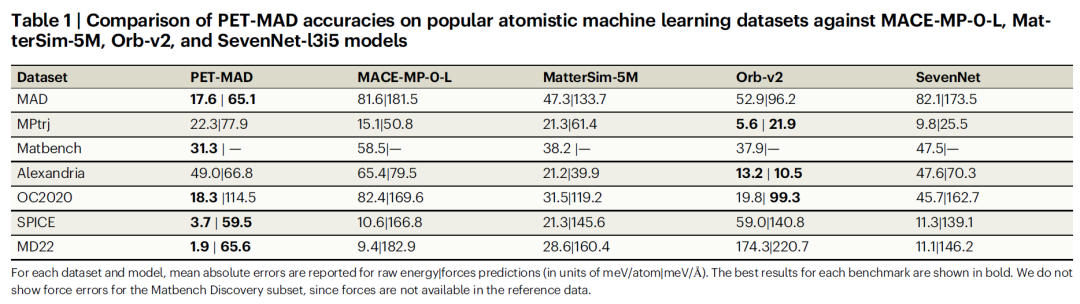
Um die Leistungsfähigkeit von PET-MAD umfassend zu bewerten, verglich das Forschungsteam es zunächst mit gängigen Modellsystemen wie MACE-MP-0 und Orb-v2 in maßgeblichen Benchmark-Tests wie Matbench Discovery. Um faire Vergleiche zu gewährleisten, berechneten die Forscher die Benchmark-Teilmenge mit PET-MAD-kompatiblen DFT-Parametern neu, um Verzerrungen durch unterschiedliche Berechnungsmethoden zu eliminieren.
Die Testergebnisse sind in der folgenden Tabelle aufgeführt.PET-MAD, trainiert mit weniger als 100.000 Strukturen (1-3 Größenordnungen weniger Daten als bei ähnlichen Modellen) und mit einer moderaten Anzahl von 2,8 Millionen Parametern (weit weniger als die 25 Millionen von Orb-v2 und die 15,8 Millionen von MACE-MP-0-L), übertrifft alle vergleichbaren Modelle auf molekularen Datensätzen (SPICE, MD22).Es ist vergleichbar mit modernen Modellen wie MatterSim-5M bei anorganischen Datensätzen, seine Vorteile sind jedoch bei Datensätzen mit stark verzerrten Konfigurationen noch deutlicher, wobei einige Modelle einen Fehler aufweisen, der bis zu 50 Mal so hoch ist wie der von MatterSim-5M.
Benchmark-Tests reichen jedoch nicht aus, um die Zuverlässigkeit des Modells in komplexen Szenarien vollständig zu überprüfen. Daher wählte das Forschungsteam sechs unterschiedliche und anspruchsvolle Anwendungsfälle aus, um die Leistung von PET-MAD, seinem LoRA-Feinabstimmungsmodell und dem speziell entwickelten Modell PET-Bespoke zu vergleichen.
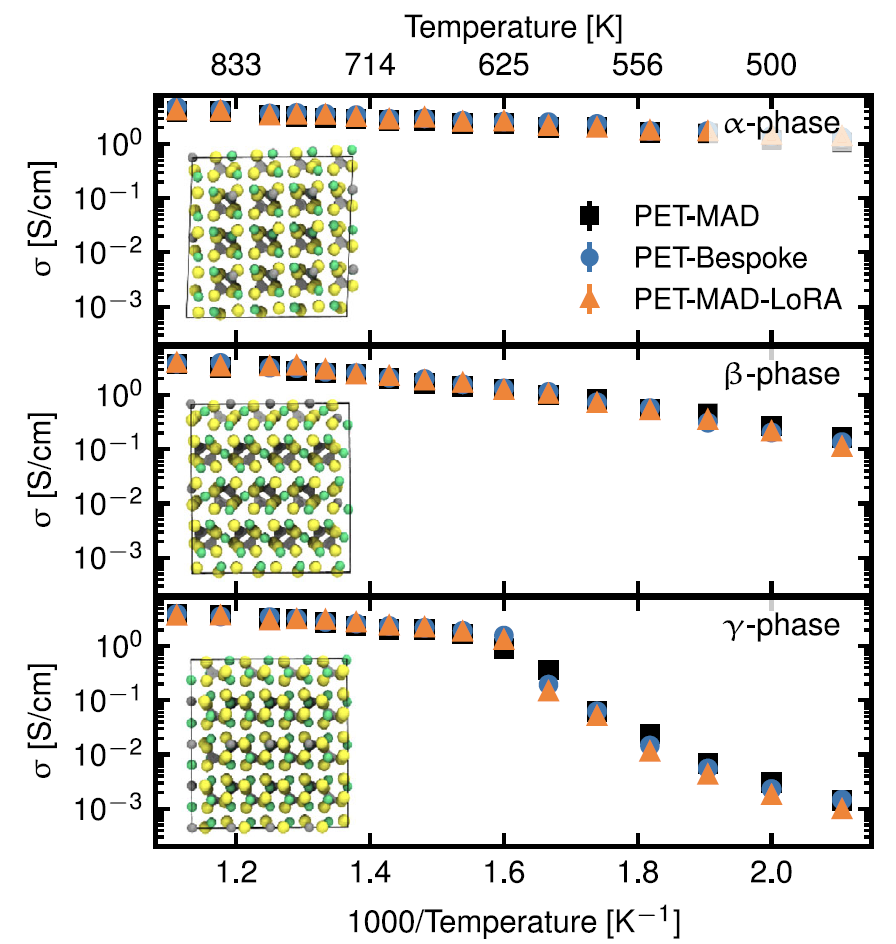
1. Festelektrolyt: Lithiumthiophosphat (LPS)
Die Studie konzentriert sich auf die wichtigste Leistungskennzahl, die Ionenleitfähigkeit. Wie die Abbildung unten zeigt, sind die Verifizierungsfehler von PET-MAD in Bezug auf Energie und Kraft (4,9 meV/Atom, 163,9 meV/Å) zwar höher als die des speziell entwickelten PET-Modells (1,2 meV/Atom, 35,6 meV/Å).Die von den Molekulardynamiksimulationen vorhergesagte Leitfähigkeit stimmt jedoch über einen weiten Temperaturbereich sehr gut mit den Ergebnissen des speziell entwickelten PET-Modells und des feinabgestimmten Modells überein.Es erfasste den Einfluss struktureller Motive (PS₄-Tetraederrotation) auf den Ionentransport präzise, wobei lediglich die Phasenübergangstemperatur der γ-Phase leicht überschätzt wurde.
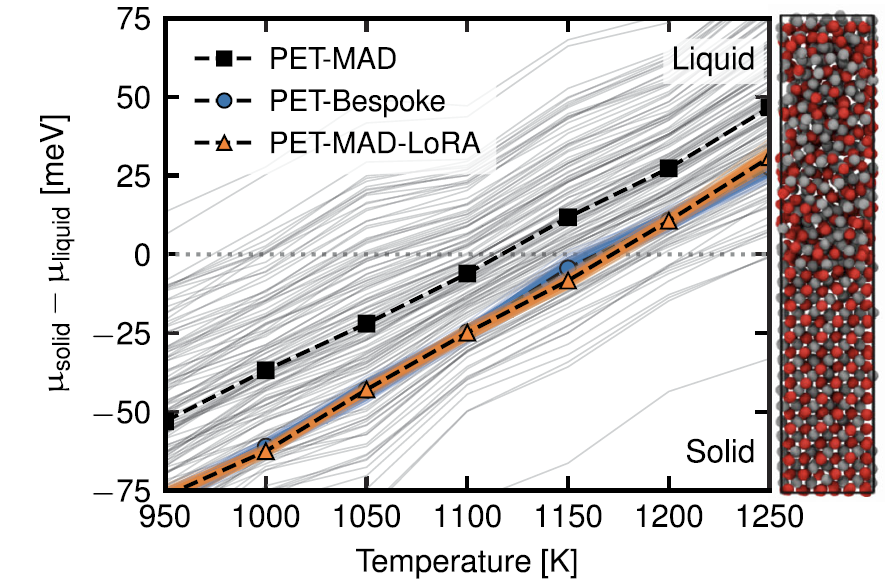
2. Halbleitermaterial: Galliumarsenid (GaAs)
Die Ergebnisse der Schmelzpunktberechnung mittels der Grenzflächenpinning-Methode sind in der folgenden Abbildung dargestellt. Der LoRA-korrigierte PET-MAD-Wert stimmt perfekt mit den Vorhersageergebnissen des speziell entwickelten PET-Modells überein (1169 ± 3 K vs. 1169 ± 4 K).
Obwohl der vorhergesagte Wert des vortrainierten allgemeinen Modells (1111±72 K) etwas niedriger ist, ist sein Fehler viel kleiner als die systembedingte Abweichung des DFT-Funktionals selbst vom experimentellen Wert (1511 K).Dieser Fall unterstreicht auch den Wert der in PET-MAD integrierten LLPR-Unsicherheitsquantifizierungsfunktion, mit der Fehler kostengünstig erfasst und übermittelt werden können und die somit einen Referenzwert für die Glaubwürdigkeit der Ergebnisse liefert.
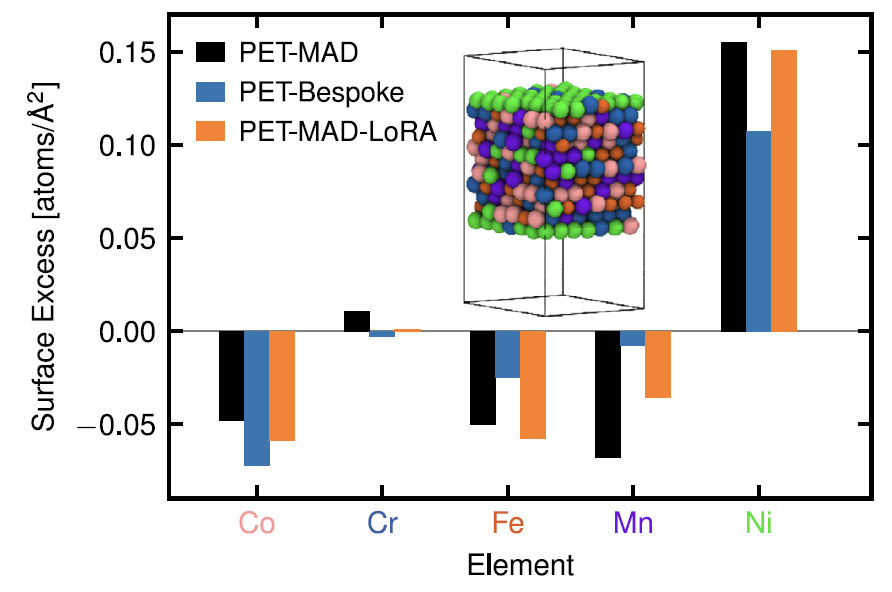
3. Hochentropielegierungen: CoCrFeMnNi
Hinsichtlich des Oberflächenentmischungsverhaltens stellte die Studie fest, wie in der folgenden Abbildung dargestellt, dass das vortrainierte PET-MAD die Entmischungseigenschaften von „nickelreicher Oberfläche und verarmten anderen Elementen“ ohne spezielles Training für dieses System genau reproduzieren kann, was in quantitativer Übereinstimmung mit der Referenzstudie steht.
Obwohl sein Energiefehler (25,8 meV/Atom) höher ist als der des speziell angefertigten PET-Modells (14,6 meV/Atom).Allerdings besteht bei letzterem die Gefahr der Überanpassung aufgrund begrenzter Trainingsdaten (nur 2000 Strukturen), während PET-MAD eine bessere Robustheit aufweist.
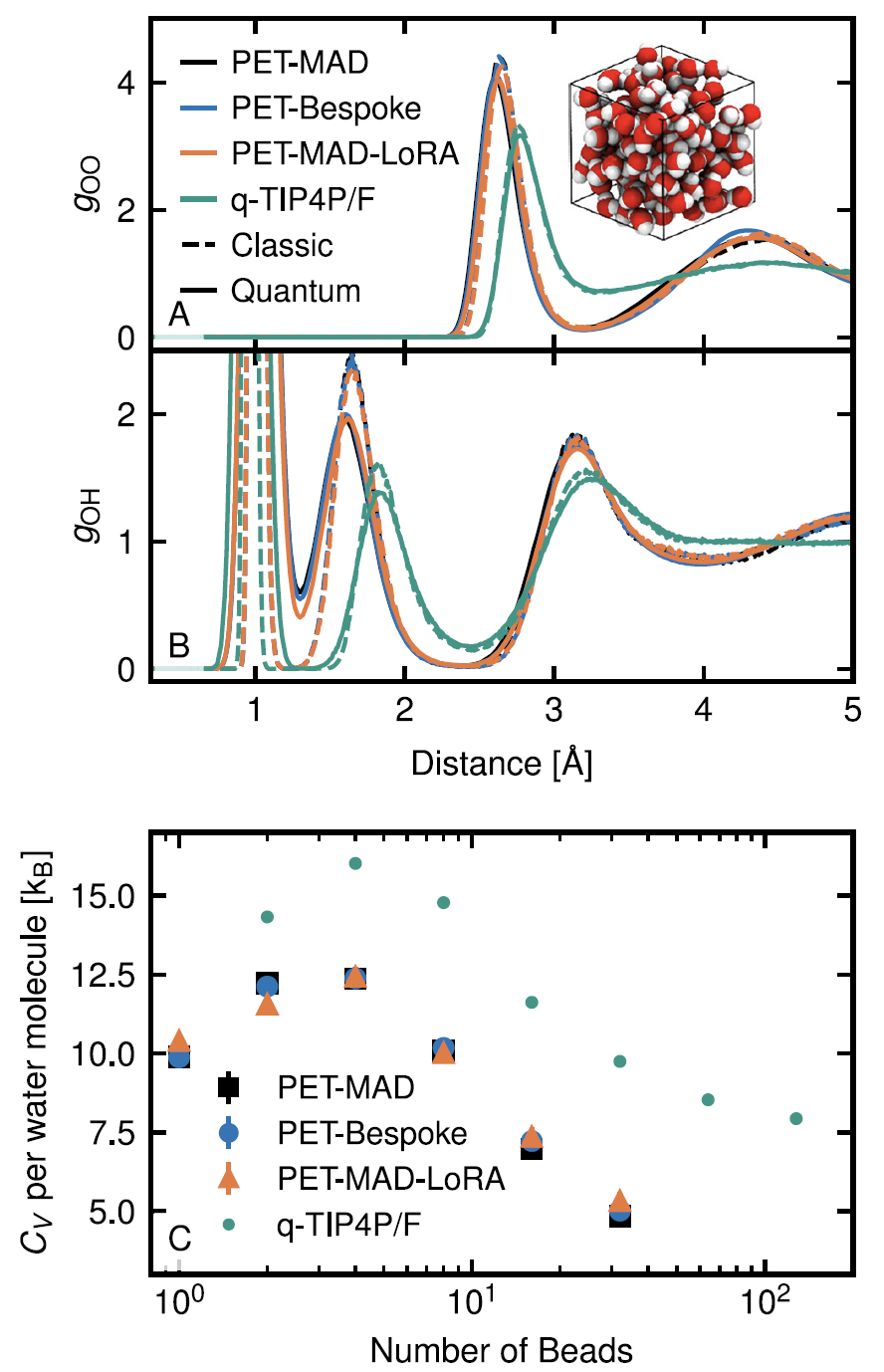
4. Flüssiges Wasser und der quantenmechanische Kerneffekt
Mithilfe von Pfadintegral-Molekulardynamiksimulationen charakterisierte PET-MAD erfolgreich die durch Quantenfluktuationen der Wasserstoffatome verursachten Phänomene der Strukturordnung und Protonendelokalisierung. Obwohl die Genauigkeit des verwendeten GGA-Funktionals begrenzt ist, zeigt das simulierte System bei Raumtemperatur Eigenschaften einer „unterkühlten Flüssigkeit“ und liefert gute Ergebnisse bei der Berechnung der radialen Verteilungsfunktion und der Wärmekapazität.Die Ergebnisse stimmen weitgehend mit dem speziell entwickelten PET-Bespoke-Modell überein, und seine Effizienz macht diese Art von rechenintensiver Quantensimulation praktikabler.
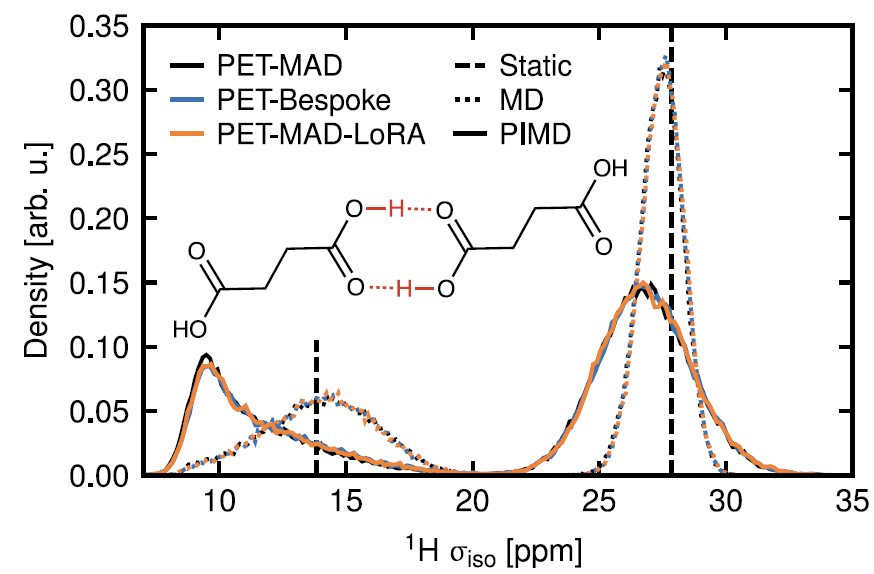
5. Molekülkristalle: NMR-Studie der Bernsteinsäure
Mithilfe von PET-MAD-gestützten kinetischen Simulationen und einem chemischen Abschirmungsmodell konnte der Einfluss der Kernquantenbewegung auf die Verteilung der ¹H-NMR-Abschirmungskonstante erfolgreich aufgezeigt werden. Wie in der Abbildung unten dargestellt, verbreitert sich die Abschirmungsverteilung wasserstoffbrückengebundener Protonen unter Berücksichtigung der Quantenabtastung deutlich und verschiebt sich zu niedrigeren Werten.Dies steht im Einklang mit den Schlussfolgerungen des speziell für PET-Bespoke entwickelten Modells, das auf Basis hochpräziser DFT-Daten trainiert wurde, und demonstriert das Potenzial von PET-MAD bei der Vorhersage komplexer funktioneller Eigenschaften.
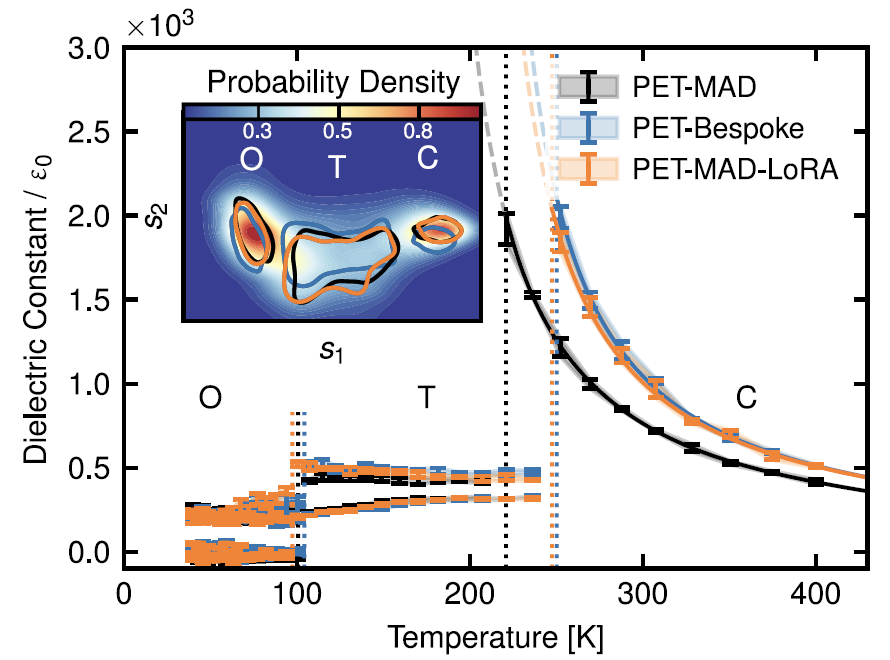
6. Ferroelektrische Materialien: Bariumtitanat (BaTiO₃)
Wie in der Abbildung unten dargestellt, reproduzierte PET-MAD in flexiblen Zellsimulationen im Temperaturbereich von 40–400 K die charakteristische Phasenübergangssequenz des Materials von rhomboedrisch über orthorhombisch und tetragonal zu kubisch präzise. Die Möglichkeiten sind jedoch durch die Dichtefunktionaltheorie (DFT) begrenzt.Die vorhergesagte Phasenübergangstemperatur ist niedriger als der experimentelle Wert, aber ihre Abweichung vom Ergebnis des speziell für PET-Bespoke entwickelten Modells beträgt weniger als 30 K.Bei der Berechnung der statischen Dielektrizitätskonstante sagte sie auch den Trend einer hohen Dielektrizitätskonstante in der kubischen Phase und einer niedrigen Dielektrizitätskonstante in der ferroelektrischen Phase korrekt voraus.
Sowohl Benchmark-Tests als auch Anwendungen in multivariaten Systemen zeigen, dass...Mit PET-MAD lässt sich eine Simulationsgenauigkeit erzielen, die nahezu der von PET-Bespoke-spezifischen Modellen entspricht, und zwar mit sehr wenigen proprietären Daten. Eigenschaften wie Ionentransport, Phasenübergänge und Oberflächenreaktionen können zuverlässig vorhergesagt werden.Durch die LoRA-Feinabstimmung kann die Leistung des speziell für PET entwickelte Modells weiter angenähert werden, während die LLPR-Fehlerabschätzung und der Dual-Force-Prediction-Modus die Effizienz und Robustheit komplexer Simulationen gewährleisten und es somit zu einem vielseitigen Werkzeug für interatomare Potentiale mit sowohl Breite als auch Tiefe machen.
MLIPs: Die Integration von Industrie, Wissenschaft und Forschung treibt einen Paradigmenwechsel in der Materialforschung und -entwicklung voran.
Dank ihrer signifikanten Vorteile wie hoher Präzision und Effizienz entwickelt sich die MLIP-Forschung zunehmend zu einer Schlüsselverbindung zwischen modernster quantenchemischer Theorie und industriellen Anwendungen. In den letzten Jahren hat sich der Forschungsschwerpunkt von der Überprüfung der Machbarkeit von Methoden hin zur Erweiterung ihrer Leistungsfähigkeit verlagert.
Ein Forschungsteam der Universität Cambridge und der Eidgenössischen Technischen Hochschule Lausanne (EPFL) kombinierte beispielsweise aktives Lernen mit erweiterten Stichprobenstrategien.Dadurch kann das Modell selbstständig wichtige Übergangszustandskonfigurationen entlang chemischer Reaktionswege untersuchen und so genaue Vorhersagen der Katalysatoraktivität und -selektivität erzielen, ohne dass umfangreiche vorherige Reaktionsdaten erforderlich sind.Es überwindet effektiv die Einschränkungen bestehender MLIPs bei der Beschreibung der dynamischen Prozesse chemischer Reaktionen.
Titel der Studie: Aedes aegypti Argonaute 2 kontrolliert Arbovirusinfektion und Wirtssterblichkeit
Link zum Artikel:
https://www.nature.com/articles/s41467-023-41370-y
Die systematische Evaluierung und die mechanistische Aufklärung bestehender Modelle werden unterdessen kontinuierlich vertieft. Das Team der University of California führte eine umfassende Evaluierung gängiger allgemeiner MLIP-Modelle wie M3GNet, CHGNet und MACE-MP-0 durch.Es umfasst eine Reihe von Materialeigenschaften, darunter Oberflächenenergie, Defektbildungsenergie, Phononenspektrum und Ionenmigrationsbarriere.Die Studie ergab, dass diese Modelle im Allgemeinen eine systematische Unterschätzung von Energie und Kraft bei Aufgaben mit hochenergetischen Zuständen oder Nichtgleichgewichtskonfigurationen aufweisen, was den Weg für weitere Verbesserungen bei den Modelltrainingsstrategien und der Datenkonstruktion weist.
Titel der Arbeit: Systematische Weichmachung in universellen interatomaren Potentialen des maschinellen Lernens
Link zum Artikel:
https://www.nature.com/articles/s41524-024-01500-6
Zusammenfassend lässt sich sagen, dass die Entwicklung des MLIP-Feldes eine neue Phase erreicht hat: von algorithmischer Innovation hin zu problemorientierten Ansätzen und von grundlegender Evaluierung hin zur Entwicklung detaillierter Anwendungsszenarien. Zukünftig, mit der Bewältigung weiterer Herausforderungen der skalenübergreifenden Modellierung und der Weiterentwicklung branchenspezifischer Modelle, werden MLIPs voraussichtlich eine zentralere Rolle im digitalen F&E-System spielen und die gegenseitige Stärkung von Materialwissenschaft und industrieller Innovation beschleunigen.








