Command Palette
Search for a command to run...
Die Modellparameter Übersteigen Die RF-Diffusion Um Das Fünffache! NVIDIA Und Andere Veröffentlichen Proteina, Ein Von Grund Auf Neu Entwickeltes Protein-Backbone Mit SOTA-Leistung

Seit dem letzten Jahrhundert widmen sich Wissenschaftler der Erforschung der Vorhersage von Proteinstrukturen auf der Grundlage von Aminosäuresequenzen. Dabei verfolgen sie die Vision, mithilfe von Aminosäuren neue Proteine zu erzeugen und den Bauplan des Lebens zu erstellen. Allerdings kam diese große Mission im Laufe der Zeit nur langsam voran. Erst in den letzten Jahren erhielt die KI-Technologie mit ihrer rasanten Entwicklung einen starken Impuls und gelangte auf die Überholspur der Entwicklung.
Seit 2016 verändert eine von Xu Jinbo, dem Gründer und Chefwissenschaftler von Molecular Heart, und anderen initiierte technologische Revolution dieses Feld im Stillen. Sie waren Pioniere bei der Einführung der ResNet-Architektur (Deep Residual Network) in den Bereich der Strukturvorhersage.Es gelang, eine signifikante Verbesserung der Proteinrückstandskontaktvorhersage zu erzielen.Dieser Durchbruch hat eine solide Grundlage für die tiefe Integration von KI und Proteindesign gelegt. Seitdem sind viele wissenschaftliche Forschungsteams diesem Beispiel gefolgt und haben hart auf diesem Gebiet gearbeitet. Es sind zahlreiche Algorithmen entstanden, die Koevolution und Deep Learning kombinieren. Darunter sind eine Reihe bedeutender Errungenschaften von David Baker, dem Nobelpreisträger für Chemie 2024, und AlphaFold, die noch bekannter geworden sind und die Forschung auf diesem Gebiet auf ein neues Niveau gehoben haben.
Bei der Überprüfung früherer StudienModelle zur bedingungslosen Generierung von Proteinstrukturen werden häufig nur anhand kleiner Datensätze trainiert, wobei die Anzahl der Strukturen 500.000 nicht übersteigt.Darüber hinaus mangelt es den neuronalen Netzwerken dieser Modelle während des Syntheseprozesses an effektiven Kontrollmethoden und es besteht eine große Lücke sowohl hinsichtlich des Umfangs als auch der Leistung im Vergleich zu generativen Modellen in Bereichen wie der natürlichen Sprache sowie der Bild- oder Videogenerierung.
Auf dem Gebiet der natürlichen Sprach-, Bild- und Videogenerierung wurden die Menschen Zeugen bahnbrechender Veränderungen und bedeutender Durchbrüche, die durch skalierbare neuronale Netzwerkarchitekturen, umfangreiche Trainingsdatensätze und eine feine semantische Kontrolle ermöglicht wurden. Dies brachte die Forscher zum Nachdenken: Können wir aus den erfolgreichen Erfahrungen in diesen Bereichen lernen und ähnliche Erweiterungen und Kontrollen an Proteinstrukturdiffusions- und -flussmodellen vornehmen, um so einen qualitativen Sprung im Bereich des Proteindesigns zu erreichen?
Erfreulich ist, dass NVIDIA kürzlich mit Mila, dem Quebec Artificial Intelligence Research Institute, der Universität Montreal und dem Massachusetts Institute of Technology zusammengearbeitet hat, um einen neuen Typ eines groß angelegten Protein-Backbone-Generators namens Proteina zu entwickeln. Proteina verfügt über die fünffache Anzahl an Parametern des RFdiffusion-Modells und erweitert die Trainingsdaten auf 21 Millionen synthetische Proteinstrukturen.Wir erreichen Spitzenleistung beim De-novo-Design von Proteinrückgraten und erzeugen vielfältige und gestaltbare Proteine von beispielloser Länge – bis zu 800 Resten.
Die entsprechenden Forschungsergebnisse mit dem Titel „Proteina: Scaling Flow-based Protein Structure Generative Models“ wurden für ICLR 2025 Oral ausgewählt.

Papieradresse:
https://openreview.net/forum?id=TVQLu34bdw&nesting=2&sort=date-desc
Empfehlen Sie eine Veranstaltung zum wissenschaftlichen Austausch. Die letzte Einladung zur Live-Übertragung von Meet AI4S erfolgt am 7. März um 12:00 Uhr.Huang Hong, außerordentlicher Professor an der Huazhong University of Science and Technology, Zhou Dongzhan, Nachwuchsforscher am AI for Science Center des Shanghai Artificial Intelligence Laboratory, und Zhou Bingxin, Assistenzforscher am Institute of Natural Sciences der Shanghai Jiao Tong University,Stellen Sie persönliche Erfolge vor und teilen Sie Erfahrungen aus der wissenschaftlichen Forschung.
KI ermöglicht Proteindesign: von der Struktur zur Sequenz, von der Vorhersage zum Design
Im Verlauf der biowissenschaftlichen Forschung hat das Proteindesign schon immer eine äußerst wichtige Rolle gespielt. Das Erlernen von Regeln und Mustern aus riesigen Mengen von Proteinsequenzdaten war für wissenschaftliche Forscher lange Zeit eine Herausforderung. Glücklicherweise hat dieser Bereich mit Unterstützung der KI-Technologie die Führung bei der Wende übernommen.
Zum Beispiel,Das von DeepMind eingeführte AlphaFold3 verbessert die Modellierung von DNA-, RNA- und kleinen Molekülinteraktionen.Die Fähigkeit, die Struktur von Proteinkomplexen genau vorherzusagen, trägt wesentlich zum Verständnis der komplexen Wechselwirkungen von Proteinen innerhalb von Zellen bei. Meta hat einmal ESMFold eingeführt, das Sprachmodell mit Strukturvorhersage kombiniert.Dadurch wird die Vorhersagegeschwindigkeit erheblich verbessert, sodass Forscher effizienter Informationen zur Proteinstruktur erhalten können.Microsofts neuestes BioEMU-1 simuliert die dynamischen Änderungen der Proteinkonformation.Es hat einen neuen Weg für die eingehende Erforschung des Bewegungsmechanismus von Proteinen und der Arzneimittelentwicklung eröffnet.
Auf dieser Grundlage hat die KI allmählich begonnen, in den Entwurf von Proteinstrukturen einzudringen.
Das Design von Proteinstrukturen basiert hauptsächlich auf bekannten Proteinstrukturen, die durch verschiedene Methoden modifiziert und optimiert werden, um Proteine mit spezifischen Funktionen oder Eigenschaften zu erhalten. Da die Funktion eines Proteins hauptsächlich durch seine dreidimensionale Konformation bestimmt wird,Die Methode der direkten Modellierung der Strukturverteilung hat sich allmählich zum Mainstream-Trend entwickelt, wobei Algorithmen, die auf Diffusionsmodellen oder Strömungsmodellen basieren, besonders hervorstechen.Beispielsweise ist das von Generate Bio entwickelte Chroma-Modell die erste groß angelegte Anwendung der Diffusionsmodellierung für ein präzises Proteindesign.Kann „Proteine produzieren, die in der Natur überhaupt nicht vorkommen“.
Auch,Die von David Baker vorgeschlagene RFdiffusion kann durch Feinabstimmung des Strukturvorhersagenetzwerks RoseTTAFold Proteinskelette mit spezifischen Funktionen erzeugen.Es bietet eine präzise strukturelle Grundlage für die Entwicklung funktioneller Proteine. Das von Forschern der Columbia University und der Rutgers University vorgeschlagene Genie2 erweitert die Trainingsdaten auf AFDB.Kann komplexe Proteine mit mehreren unabhängigen Funktionsstellen erzeugen.
Es ist bekannt, dass Struktur und Sequenz von Proteinen miteinander verbunden sind, die Struktur die Funktion bestimmt und die Sequenz die Grundlage der Struktur bildet. Wenn die Proteinstruktur durch KI-Technologie verändert wird, ändert sich zwangsläufig auch die Proteinsequenz. Das Design von Proteinsequenzen basiert hauptsächlich auf der bekannten Proteinstruktur und basiert auf Berechnungs- und Vorhersagemethoden, um eine Aminosäuresequenz zu entwerfen, die der Struktur entspricht.
Derzeit wird das AI-Proteinsequenzdesign hauptsächlich in zwei Typen unterteilt:Eines davon ist ein Tool zur Entwicklung einer Proteinsequenz mit festem Rückgrat,Beispielsweise verwendet das von der Stanford University eingeführte ESM-IF ein Paradigma, das Vortraining und Feinabstimmung kombiniert und so Strukturwissen geschickt in das funktionelle Proteindesign integriert, wodurch das Design von Proteinen mit spezifischen Funktionen stark unterstützt wird. Das von David Baker vorgeschlagene ProteinMPNN basiert auf einem Graph-Neural-Network und kann passende Aminosäuresequenzen entsprechend der Hauptkettenstruktur generieren und bietet so eine effiziente und genaue Methode für das Protein-Sequenz-Design.
Das andere ist ein funktionsorientiertes Tool zum Design von Proteinsequenzen.Beispielsweise ist ProGen von Salesforce ein bedingtes Generierungsmodell, das Proteinsequenzen entsprechend spezifischer funktionaler Anforderungen anpassen kann und so eine äußerst flexible Lösung für die Entwicklung funktionaler Proteine bietet. ZymCTRL, das von der Universität Girona in Spanien eingeführt wurde, erreicht funktionsorientiertes Design durch Feinabstimmung des vortrainierten Sprachmodells und bietet so starke Unterstützung für die präzise Regulierung der Proteinfunktion. Die vom Tianjin Institute of Industrial Biotechnology der Chinesischen Akademie der Wissenschaften vorgeschlagene P450Diffusion erzeugt P450-Enzymvarianten mit spezifischen katalytischen Funktionen auf der Grundlage des Diffusionsmodells und eröffnet so neue Entwicklungsmöglichkeiten im Bereich der Enzymtechnik.
Im Vergleich zu den anderen drei Arten von Proteinmodellen ist der Maßstab aktueller Proteinstrukturdesignmodelle jedoch im Allgemeinen klein. Insbesondere liegt die Trainingssatzgröße von AlphaFold 3 bei fast 100 Millionen, BioEmu-1 verwendet in der Vortrainingsphase mehr als 200 Millionen Proteinsequenzen aus der AFDB-Datenbank und die Anzahl der Parameter von ProGen beträgt bis zu 1,2 Milliarden. RFdiffusion, ein herausragender Vertreter auf dem Gebiet des Proteinstrukturdesigns, verwendet seine Trainingsdaten jedoch nur aus Zehntausenden realer Proteinstrukturen im Protein Data Bank (PDB)-Repository, und die Gesamtlänge der von ihm erstellten Struktur kann nur 600 Aminosäurereste erreichen. Der größte Datensatz von Genie2 umfasst nur etwa 600.000 synthetische Strukturproteine.
In diesem ZusammenhangDie Branche sieht der Geburt eines Proteinstruktur-Designmodells mit größerem Trainingsdatenvolumen, längerer Gesamtstrukturlänge und stärkerer Steuerbarkeit mit Spannung entgegen – Proteina.
Proteina-Modell: Ein neuer Durchbruch im Proteindesign mithilfe von KI-Technologie
Proteina verwendet als flussbasiertes Proteinstrukturmodell eine innovative skalierbare nicht-äquivariante Transformer-Architektur, die vom Diffusions-Transformer im Sichtfeld inspiriert ist. Es kann Spitzenleistungen erzielen, auch ohne auf die rechenintensive Dreiecksschicht zurückzugreifen.Dadurch kann Proteina anhand von bis zu 21 Millionen Proteinstrukturen trainiert werden, was einer 35-fachen Steigerung der Trainingsdaten entspricht und letztendlich Backbones mit bis zu 800 Resten erzeugt.Unter Beibehaltung der Gestaltbarkeit und Vielfalt ist es deutlich besser als alle bisherigen Arbeiten.
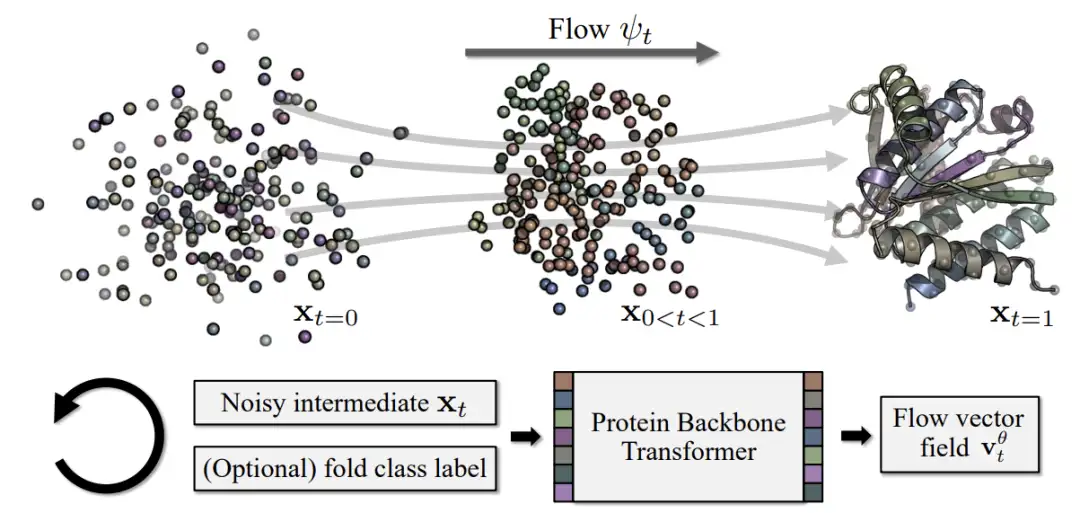
Wie in der folgenden Abbildung gezeigt, verwendet diese Studie hauptsächlich den von Genie2 verwendeten Foldseek AFDB-Clustering-DFS-Datensatz.Der Datensatz umfasst etwa 600.000 synthetische Strukturproteine.Gleichzeitig wurde in der Studie auch die hochwertig gefilterte AFDB-Teilmenge D21M verwendet, die aus etwa 214 Millionen AFDB-Strukturen gefiltert wurde.Diese Untergruppe enthält ungefähr 21 Millionen synthetische Strukturproteine.
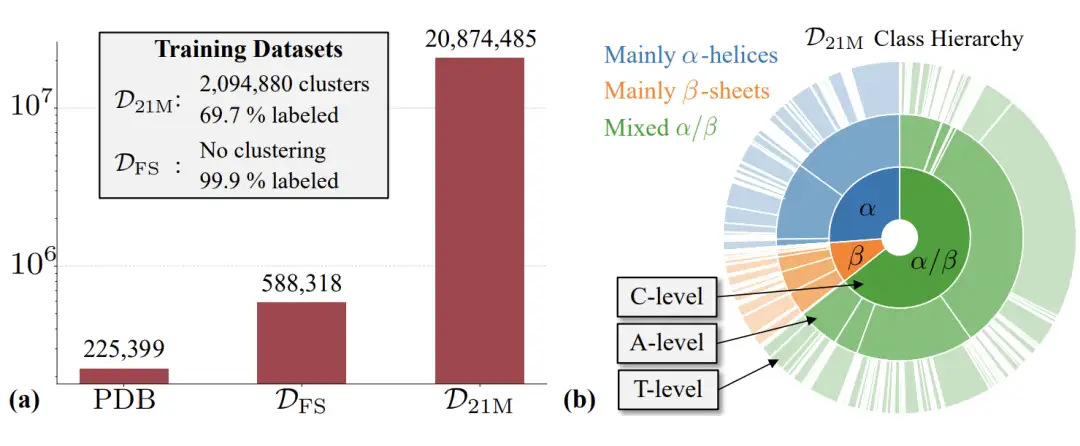
Basierend auf den beiden oben genannten Datensätzen trainierten die Forscher drei weitere Proteina-Modelle: Das erste ist das MFS-Modell, das einen Transformer mit 200 Millionen Parametern und eine Dreiecksschicht mit 10 Millionen Parametern enthält; Das zweite ist das Mno-triFS-Modell, das nur einen Transformer mit 200 Millionen Parametern enthält, aber keine Aktualisierungen der Dreiecksschicht oder paarweisen Darstellung enthält. Das dritte ist das M21M-Modell, das einen Transformer mit 400 Millionen Parametern und eine Dreiecksschicht mit 15 Millionen Parametern enthält.
Im Bereich der Generierung unbedingter Proteinstrukturen haben lange Zeit isovariante Methoden dominiert, doch Proteina zeigt, dass auch groß angelegte nicht-isovariante Flussmodelle erfolgreich sein können. Die Parameter seiner Trainingsversion übersteigen 400 Millionen.Es ist mehr als fünfmal größer als RFdiffusion und derzeit der größte Protein-Rückgratgenerator.Die Ergebnisse zeigen auch, dass mit DFS trainierte Modelle eine höhere Diversität aufweisen, Forscher aber auch viel größere Mengen qualitativ hochwertiger Daten aus vollständig synthetischen Strukturen erstellen können als mit DFS.
In Bezug auf Bewertungsindikatoren gibt sich Proteina nicht mit den traditionellen Bewertungen der Vielfalt, Neuheit und Gestaltbarkeit zufrieden, sondern führt innovative Bewertungsindikatoren ein – indem die empirischen Bezeichnungen von DFS direkt in das Modell eingegeben werden. Dieser Schritt erzwingt die Vielfalt zwischen verschiedenen Faltungsstrukturen und ermöglicht durch neuartige Faltungsklassenbeschränkungen eine beispiellose Kontrolle über synthetische Proteinstrukturen.
Wie in der folgenden Abbildung gezeigt, erreicht das bedingte Modell von Proteina im Vergleich zur bedingungslosen Generierung die fortschrittlichste TM-Score-Diversität und gleichzeitig die besten FPSD-, fS- und fJSD-Werte.Dies zeigt voll und ganz seinen Vorteil bei der Faltstrukturdiversität „fS“ und der besseren Verteilungsübereinstimmung zwischen der generierten Struktur und den Referenzdaten.
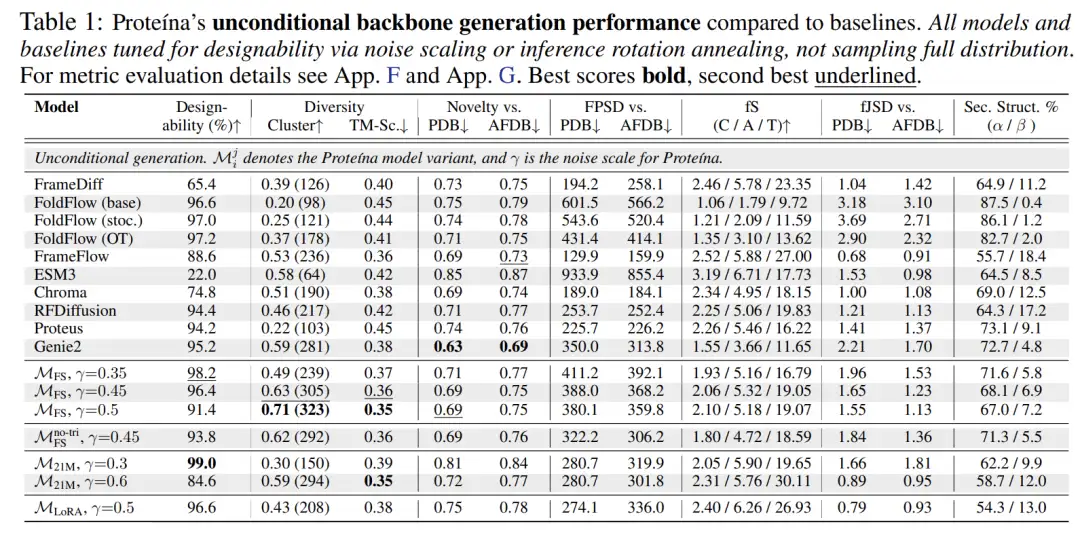
Darüber hinaus hat Proteina das Flow-Matching-Ziel angepasst, um die Generierung von Proteinstrukturen zu ermöglichen, und stufenweise Trainingsstrategien erforscht, wie etwa die Verwendung von LoRA zur Feinabstimmung des Modells, um ihm die Generierung natürlicher, gestaltbarer Proteine zu ermöglichen. Darüber hinaus wurden neue Leitschemata für bedingte Beschränkungen hierarchischer Faltungskategorien entwickelt und eine Selbstführung zur Verbesserung der Gestaltbarkeit von Proteinen erfolgreich demonstriert.In Bezug auf die Leistung bei der Proteinrückgratbildung hat Proteina das SOTA-Niveau erreicht, insbesondere bei der Langkettensynthese.Es übertrifft alle Basismodelle deutlich und weist durch neuartige Faltungskategorie-Bedingungsbeschränkungen bessere Steuerungsmöglichkeiten als frühere Modelle auf.
Innovationen im chinesischen Bereich des KI-Proteindesigns
Da DeepSeek derzeit erneut das große Sprachmodell entfacht, wird das Feld des Proteindesigns zweifellos neue Entwicklungsmöglichkeiten eröffnen und immer mehr chinesische Kräfte werden auftauchen. Tatsächlich haben chinesische Forscher und Unternehmen allein auf dem Gebiet des Proteinstrukturdesigns bereits zahlreiche Ergebnisse erzielt.
Im Jahr 2022Shanghai Tianrang XLab hat mithilfe künstlicher Intelligenz eine neue Proteindesign-Plattform eingeführt: TRDesign. TRDesign kann alle potenziellen Möglichkeiten im Bereich der Proteinfaltung genau erkunden, indem es viel über die Beziehung zwischen Proteinsequenz und -struktur lernt, die aus der Proteinfaltung gewonnene Sequenz-Struktur-Funktions-Assoziation umgekehrt abbildet und ein durchgängiges Proteindesign, Tests, Stabilitäts- und Affinitätsoptimierung von Grund auf durchführt und so Proteinstrukturen entwirft, die den Anforderungen besser gerecht werden.
Im Jahr 2023Professor Jinbo Xu, Gründer von Molecular Heart, stellte das große Modell von NewOrigin auf der Weltkonferenz für künstliche Intelligenz „WAIC“ 2023 vor.Durch das Lernen aus Hunderten von Milliarden multimodaler Big Data kann das Modell eine multimodale gerichtete Stromerzeugung erreichen. Ein einzelnes Modell kann alle Prozessanforderungen der Proteingenerierung erfüllen, einschließlich Sequenzgenerierung, Strukturvorhersage, Funktionsvorhersage und De-novo-Design. Es kann das Problem der Erzeugung spezifischer funktioneller Proteine lösen, die für industrielle Anwendungen erforderlich sind, und seine Auswirkungen und seinen Wert in einer realen industriellen Umgebung bewerten.
Im April 2024 schloss sich Wuxi Tushen Zhihe Artificial Intelligence Technology Co., Ltd. mit mehreren Forschungseinrichtungen zusammen, umGemeinsame Veröffentlichung von Chinas erstem großen Textproteinmodell für natürliche Sprache, TourSynbio. Das große Modell von TourSynbio öffnet den Proteindesignprozess und realisiert „Protein Design AI in One“. Es kann eine detaillierte Darstellung jedes Proteins liefern, Dialoge und Eingabeaufforderungen in natürlicher Sprache unterstützen und den Proteindesignprozess erheblich vereinfachen.
August 2024Das Team von Zhang Haicang vom Institut für Computertechnologie der Chinesischen Akademie der Wissenschaften schlug CarbonNovo vor.Dieses Ergebnis wurde in ICML2024 veröffentlicht. CarbonNovo entwirft gemeinsam die Struktur und Sequenz des Proteinrückgrats von Anfang bis Ende. Es verbessert effektiv die Designeffizienz und -leistung durch die Erstellung eines gemeinsamen Energiemodells und die Einführung eines Proteinsprachenmodells und bietet damit erhebliche Vorteile gegenüber dem bestehenden zweistufigen Designmodell.
Link zum Artikel:
https://openreview.net/pdf?id=FSxTEvuFa7 Code-Link:
https://github.com/zhanghaicang/carbonmatrix_public
Im Oktober 2024 werden das Team von Professor Liu Haiyan und Professor Chen Quan von der School of Life Sciences and Medicine des USTC,Wir haben ein Protein-Rückgrat-Rauschunterdrückungs-Diffusionswahrscheinlichkeitsmodell SCUBA-D entwickelt, das nicht auf einem vorab trainierten Strukturvorhersagenetzwerk basiert.Es kann die Hauptkettenstruktur automatisch von Grund auf neu entwerfen und so eine vollständige Toolkette bilden, mit der künstliche Proteine mit neuen Strukturen und Sequenzen von Grund auf neu entworfen werden können. Es handelt sich neben RosettaDesign um die einzige Methode zum Protein-De-novo-Design, die vollständig experimentell verifiziert wurde. Die entsprechenden Ergebnisse wurden in Nature Methods veröffentlicht.
Link zum Artikel:
https://doi.org/10.1038/s41592-024-02437-W
Im Jahr 2025 kombinierte das Team von Lu Peilong an der Westlake University Deep Learning und energiebasierte Methoden, umDas transmembranäre fluoreszenzaktivierte Protein tmFAP, das spezifisch an fluoreszierende Liganden binden kann, wurde erfolgreich entwickelt.Zur Lösung der Kernprobleme beim Design transmembranärer Proteine wurden Deep-Learning-Algorithmen eingesetzt. Zum ersten Mal gelang die präzise De-novo-Gestaltung nichtkovalenter Wechselwirkungen zwischen Transmembranproteinen und Ligandenmolekülen innerhalb der Membran. Zudem wurde ihre Fähigkeit zur Fluoreszenzaktivierung in lebenden Zellen nachgewiesen, wodurch neue Wege für die Entwicklung und Anwendung von Transmembranproteinen eröffnet wurden. Diese Forschungsergebnisse wurden in Nature, einer führenden internationalen Fachzeitschrift, veröffentlicht.
Link zum Artikel:
https://www.nature.com/articles/s41586-025-08598-8
Derzeit hat China im Bereich des KI-gesteuerten Proteindesigns ein einzigartiges technologisches Ökosystem aufgebaut. Seine bahnbrechenden Fortschritte spiegeln sich nicht nur im Innovationsgrad der Algorithmen wider, sondern auch im Aufbau einer vollständigen Innovationskette von der Grundlagentheorie bis zur industriellen Anwendung. Das Aufkommen dieser Erfolge verdeutlicht die Tiefe und Breite der technologischen Durchbrüche Chinas auf dem Gebiet des Proteindesigns. Ich bin davon überzeugt, dass es mit der kontinuierlichen Weiterentwicklung der KI-Technologie in Zukunft noch weitere bemerkenswerte Erfolge geben wird, die einen Paradigmenwechsel in der Entwicklung der weltweiten Biowissenschaftsforschung und der biopharmazeutischen Industrie mit sich bringen werden.








