Command Palette
Search for a command to run...
Die Herausforderung Der Modellierung Von Proteinkonformationsheterogenität Auf Atomarer Ebene Lösen! PLACER-Framework-Analyse Von David Bakers Team

In der molekularen Welt sind die Wechselwirkungen zwischen Proteinen und Nukleinsäuren, kleinen organischen und anorganischen Molekülen sowie Metallionen für lebenswichtige Prozesse unerlässlich. Jede Erkennung und Bindung dieser Wechselwirkungen kann biologische Funktionen beeinflussen, die Wirksamkeit von Medikamenten bestimmen und sogar über Erfolg oder Misserfolg bei der Entwicklung neuer Enzyme entscheiden. Die Modellierung dieser Wechselwirkungen auf atomarer Ebene und ihrer Konformationsheterogenität stellt die Industrie jedoch weiterhin vor große Herausforderungen.
Auf Deep Learning (DL) basierende Docking-Tools für kleine Moleküle wie DiffDock weisen eine höhere Genauigkeit im Vergleich zu früheren Methoden auf.Bei Aufgaben mit hohen Präzisionsanforderungen ist der Leistungsunterschied jedoch nicht signifikant.Darüber hinaus verschlechtert sich die Leistung erheblich beim Auftreffen auf unbekannte Rezeptoren; außerdem wurden verschiedene auf Deep Learning basierende Methoden entwickelt, um Konformationen kleiner Moleküle aus chemischen Strukturen zu generieren.Allerdings modellieren diese Methoden typischerweise nur bestimmte Kategorien interagierender Objekte, wodurch ihre Fähigkeit, das gesamte Spektrum der Proteinfunktion zu charakterisieren, eingeschränkt ist.
Darauf aufbauend entwickelte ein Forschungsteam unter der Leitung des Nobelpreisträgers Professor David Baker von der University of Washington ein Graph-Neuronales Netzwerk namens PLACER (Protein-Ligand Atomistic Conformational Ensemble Resolver).Es kann die Strukturen verschiedener organischer kleiner Moleküle präzise auf Basis der atomaren Zusammensetzung und der Bindungsinformationen der kleinen Moleküle generieren; und es kann, unter Berücksichtigung der makroskopischen Strukturumgebung von Proteinen, die detaillierten Strukturen kleiner Moleküle und Proteinseitenketten für Protein-Kleinmolekül-Docking-Aufgaben konstruieren.Im Rahmen der Enzymdesignforschung entdeckte dieses Forschungsteam, dass die Verwendung von PLACER zur Bewertung der Genauigkeit designter aktiver Zentren und des Präorganisationsgrades die Erfolgsquote des Designs und die Enzymaktivität signifikant verbessern kann. So erhielten die Forscher beispielsweise eine präorganisierte Antialdolase mit einem kcat/KM-Wert von 11.000 M⁻¹·min⁻¹.Es übertrifft bei Weitem alle Designergebnisse vor dem Aufkommen des Deep Learning.
Die zugehörigen Forschungsergebnisse mit dem Titel „Modellierung von Protein-Kleinmolekül-Konformationsensembles mit PLACER“ wurden in den Proceedings of the National Academy of Sciences (PNAS) veröffentlicht.
Forschungshighlights:
* PLACER arbeitet schnell und zufällig, wodurch es in der Lage ist, schnell eine große Anzahl von Vorhersagebeispielen zu generieren, um die Verteilung der Konformationsheterogenität darzustellen.
* Durch die Verwendung einer einheitlichen atomaren Darstellung aller Wechselwirkungen kann PLACER problemlos über Biomoleküle hinaus erweitert werden, beispielsweise auf makrozyklische Moleküle und andere komplexe kleine Moleküle.
* PLACER ist von großem Wert für das computergestützte Enzymdesign und das Design von Konjugaten kleiner Moleküle: Es kann schnell die Genauigkeit der Rekonstruktion des entworfenen aktiven Zentrums und die Vororganisation wichtiger katalytischer/interagierender Seitenkettenfunktionalgruppen beurteilen.

Papieradresse:
https://www.biorxiv.org/content/10.1101/2024.09.25.614868v2
Folgen Sie unserem offiziellen WeChat-Account und antworten Sie im Hintergrund mit „Enzymdesign“, um das vollständige PDF zu erhalten.
Datensätze: Die mehrstufige und vielfältige Datenkonstruktion bestätigt eine ausgezeichnete Generalisierungsfähigkeit
Für die Konformationsvorhersage kleiner Moleküle wählte das Team über 226.000 organische, nicht-polymere Kristallstrukturen kleiner Moleküle aus der Cambridge Structural Database (CSD) als Trainingsdatensatz und 7.116 Proben als Validierungsdatensatz aus. Jedes Molekül liefert vollständige Informationen zur atomaren Zusammensetzung und zu den chemischen Bindungen, während die Atomkoordinaten zufällig initialisiert werden. Dadurch kann das Modell lernen, auch unter verrauschten Bedingungen präzise Strukturen zu rekonstruieren.Diese Trainingsstrategie gewährleistet nicht nur, dass das Modell die subtilen Veränderungen kleiner Moleküle in verschiedenen Konformationen erfassen kann, sondern ermöglicht auch die Generierung einer vielfältigen Menge an Molekülkonformationen durch mehrere Durchläufe.
Im Hinblick auf Protein-Kleinmolekül-Systeme wählte das Forschungsteam hochauflösende Strukturen (<2,5 Å) aus der Proteindatenbank (PDB) aus, darunter Protein-Kleinmolekül-Komplexe, mit insgesamt etwa 113.000 Trainingsbeispielen und 7.090 Validierungsbeispielen. Bemerkenswert ist,Das Team schloss lediglich Wassermoleküle aus, behielt aber Informationen über potenziell nicht-biologische kleine Moleküle (wie Lösungsmittel) bei, da diese dennoch wertvolle Hinweise auf die physikochemischen Präferenzen molekularer Grenzflächen lieferten.Die Trainingsdaten werden so zugeschnitten, dass sie maximal 600 schwere Atome enthalten, und mit Gaußschem Rauschen um zufällig ausgewählte Atomzentren gestört, um die komplexe dynamische Umgebung von Proteinen und kleinen Molekülen in der Realität zu simulieren.
Diese vielschichtige und vielfältige Datenkonstruktion gewährleistet, dass PLACER eine ausgezeichnete Generalisierungsfähigkeit aufweist, wenn es um die Verarbeitung von einzelnen kleinen Molekülen bis hin zu komplexen Protein-Kleinmolekül-Systemen geht.
Das neuronale Netzwerk PLACER verwendet eine dreispurige Architektur, die sich auf Seitenketten auf atomarer Ebene und Konformationen kleiner Moleküle konzentriert.
PLACER ist ein neuronales Netzwerk zur Rauschunterdrückung, dessen Eingabe teilweise gestörte Proteinstrukturen und chemische Strukturinformationen (ohne Koordinaten) aller interagierenden Moleküle umfasst. Die Ausgabe ist die vollständige Atomstruktur des Komplexes und die Unsicherheit der Position jedes Atoms im Vorhersagemodell, wie in Abbildung A unten dargestellt.
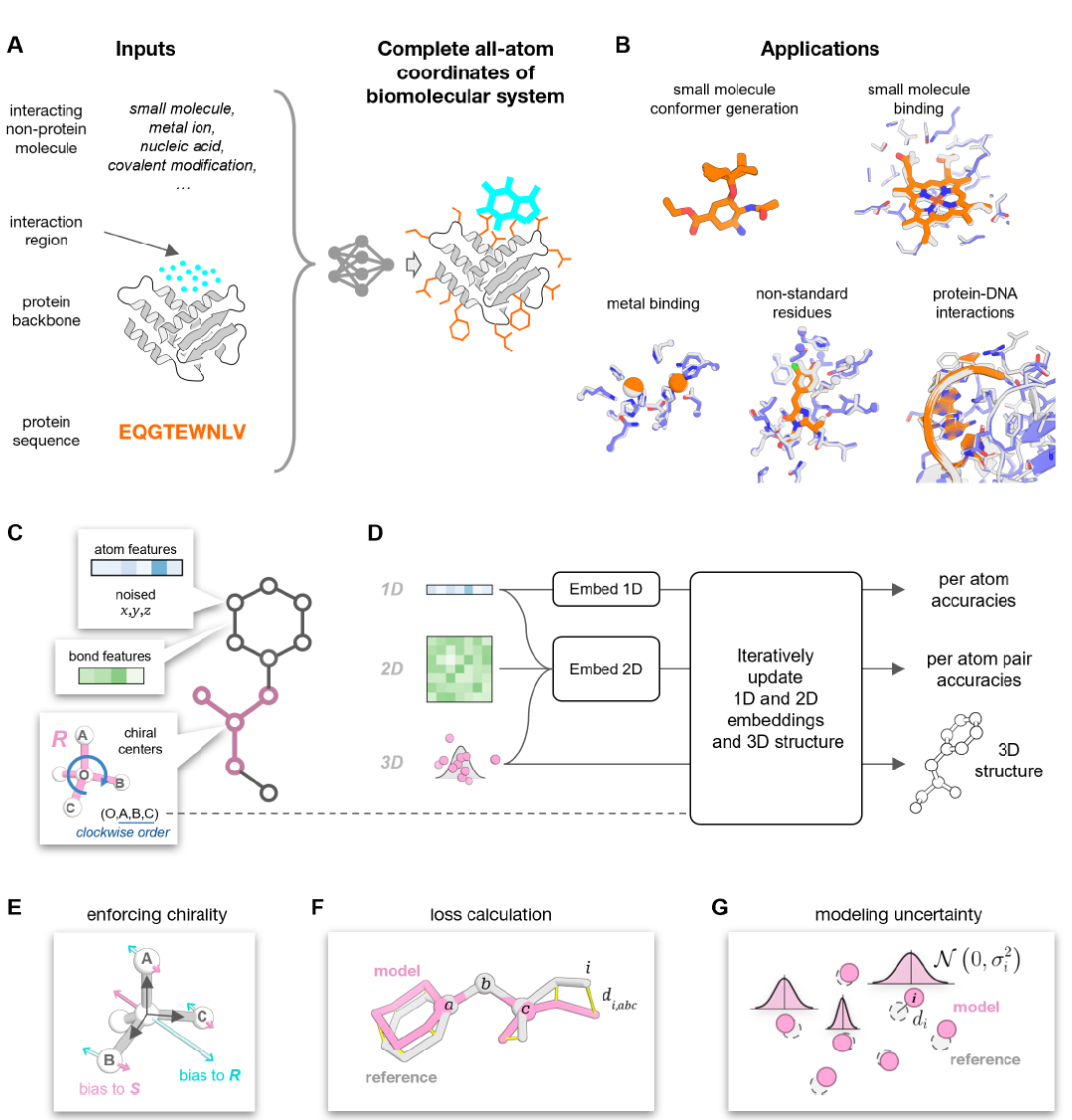
In der Eingabephase wird das Molekülsystem in einen chemischen Graphen transformiert. Die Knoten repräsentieren dabei einzelne schwere Atome (Wasserstoffatome werden zur Reduzierung des Rechenaufwands nicht modelliert), die Kanten hingegen chemische Bindungen zwischen den Atomen (siehe Abbildung C oben). Diese Darstellung ist für verschiedene Molekültypen konsistent. Jeder Knoten im Netzwerk enthält Informationen zum Atomtyp und seine anfänglich veränderten 3D-Koordinaten. Das Netzwerk hat die Aufgabe, die Eingabekoordinaten iterativ zu entrauschen und gleichzeitig die Unsicherheiten der Atompositionen in der resultierenden Modellstruktur abzuschätzen (siehe Abbildung D oben).
PLACER verwendet eine von RoseTTAFold (RF) inspirierte Drei-Spur-Architektur, und die Gesamtarchitektur des Netzwerks sieht wie folgt aus:
* Drei-Orbital-Design (1D, 2D, 3D): 1D-Orbitale verarbeiten Informationen über atomare Merkmale; 2D-Orbitale verarbeiten Beziehungen zwischen Atompaaren (wie chemische Bindungen und räumliche Nähe); 3D-Orbitale sind für die Aktualisierung der Atomkoordinaten zuständig.
* Iterative Optimierung: Nach der initialen Einbettung der 1D- und 2D-Merkmale werden diese an den Iterationsblock übergeben, um die Einbettungsvektoren und die 3D-Struktur iterativ zu aktualisieren. Im Iterationsblock wird zunächst ein atomarer Nachbarschaftsgraph erstellt – für jedes Atom werden jeweils die Hälfte der räumlichen und chemischen Nähe ausgewählt, insgesamt also 32 nächste Nachbaratome. Anschließend werden die 2D-Merkmalspaare mithilfe einer Feedforward-Adapterschicht in Kanteneinbettungen projiziert. Zusammen mit den 1D-Merkmalen, dem atomaren Nachbarschaftsgraphen und der aktuellen 3D-Atomstruktur dienen sie als Eingabe für das SE3-Transformer-Netzwerk, um die 3D-Koordinaten und die 1D-Einbettungsvektoren zu aktualisieren.
* Verarbeitung chiraler Zentren: Informationen zu chiralen Zentren werden dem Netzwerk über Typ-1-Merkmale (Vektormerkmale) zugeführt (siehe Abbildung E oben). Merkmale in 2D-Orbitalen werden paarweise aktualisiert und mit strukturellen Verzerrungen kombiniert. Die Konfidenzvorhersageköpfe für Atome und ihre Paare verzweigen sich von den 1D- bzw. 2D-Orbitalen, um die Berechnung der iterativen Blöcke abzuschließen. Das vollständig trainierte Netzwerk enthält acht iterative Blöcke mit gemeinsamen Gewichten.
* Verlustfunktionsdesign: Das PRACER-Training verwendet eine Kombination aus Strukturverlust und Konfidenzvorhersageverlust, die nach jeder Iteration angewendet werden. Der primäre Strukturverlust ist der All-Atom-FAPE (Frame-Aligned Point Error); die Konfidenz der Modellstruktur wird sowohl auf atomarer als auch auf Atompaar-Ebene bewertet.
Durch diese sorgfältig konzipierte NetzwerkarchitekturPLACER kann vielfältige und atomgenaue Sätze von Protein-Kleinmolekül-Konformationen ausgehend von zufällig initialisierten Koordinaten erzeugen.Dies bildet eine verlässliche Grundlage für nachfolgende Analysen und die Entwicklung. Im Gegensatz zu Methoden zur Vorhersage von Proteinstrukturen wie AlphaFold,PLACER sagt nicht die Hauptkettenstruktur von Proteinen voraus, sondern konzentriert sich auf Seitenketten auf atomarer Ebene und Konformationen kleiner Moleküle, wodurch die Rechengeschwindigkeit deutlich verbessert und die Generierung vielfältiger Konformationssätze ermöglicht wird.
Ergebnispräsentation: Unterstützung für Präzisionsentwicklung von kleinen Molekülen bis hin zu komplexen Proteinsystemen
Konformationsvorhersage kleiner Moleküle
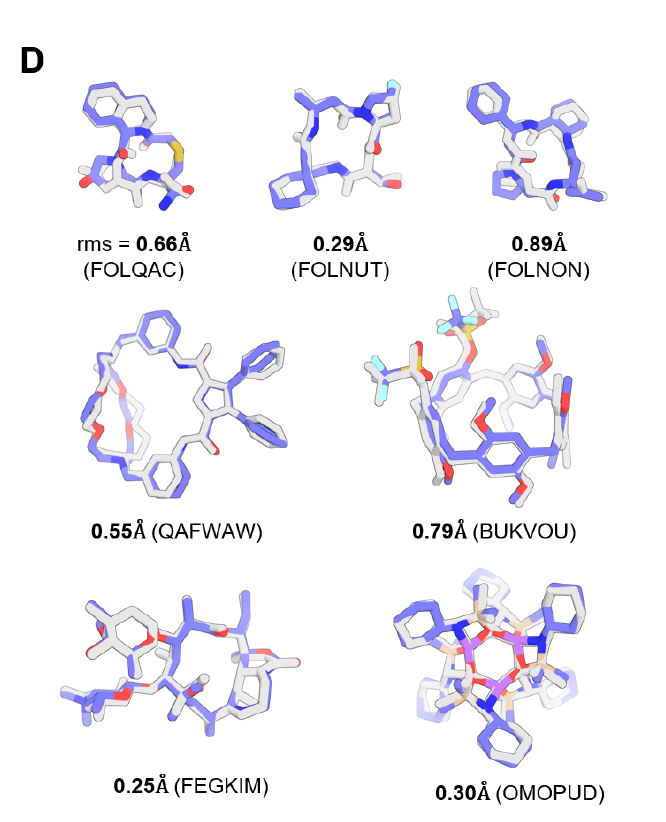
Tests mit dem CSD-Datensatz für kleine Moleküle zeigen, dass das vollständig trainierte PLACER die dreidimensionalen Strukturen komplexer Moleküle mit einer Genauigkeit im Sub-Ångström-Bereich korrekt generieren kann.Beispiele hierfür sind Makrozyklen mit mehr als 50 Atomen (siehe Abbildung D unten), einschließlich Peptidmakrozyklen (siehe obere Zeile in Abbildung D unten). Ablationsversuche zeigen, dass fehlende Informationen über Bindungsabstände oder eine Reduzierung der Iterationsanzahl die Vorhersagegenauigkeit erheblich verringern. Dies unterstreicht die entscheidende Bedeutung der für PLACER entwickelten Iterations- und Merkmalsstrategien.
Protein-Kleinmolekül-Wechselwirkungen
Mithilfe von PLACER erzeugten die Forscher eine Reihe von Konformationen kleiner Moleküle in der Tasche des Zielproteins (siehe Abbildung A unten), indem sie das Netzwerk mehrmals ausführten, jedes Mal mit unterschiedlichen zufälligen Initialisierungen der Eingabekoordinaten.Die Analyse des generierten Konformationssatzes zeigt, dass PLACER unempfindlich gegenüber der Ausgangsposition des Liganden ist: Mehrere verschiedene Startpositionen können Vorhersagen erzeugen, die nahe an der natürlichen Konformation liegen (Abbildung B unten), und diese Positionen decken den gesamten Raum der Eingabeabtastung ab (Abbildung D unten).Die Forscher stellten außerdem fest, dass der auf Basis der Ligandenatome berechnete vorhergesagte RMSD-Wert (pRMSD) dazu genutzt werden konnte, genauere Modelle aus dem Stichprobenpool auszuwählen (Abbildung E unten). Das Modell mit dem höchsten Wert zeigte eine hohe Übereinstimmung mit der experimentellen Struktur (Abbildung C unten).
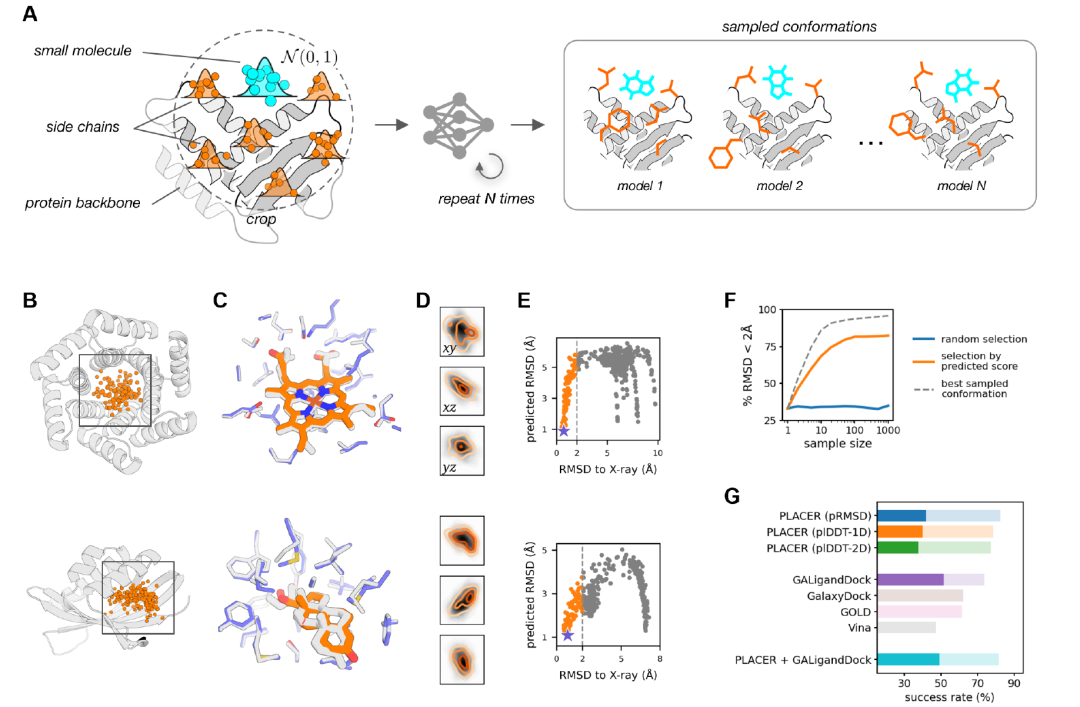
Wie in Abbildung G oben dargestellt, erzielte PLACER im Test nicht-natürlicher Konformationen von 65 Wirkstoffzielen hervorragende Ergebnisse bei der Generierung und Auswahl naturnaher Konformationen. Die Erfolgsrate, gemessen anhand des pRMSD-Scores, übertraf die von traditionellen Docking-Tools wie Vina, GOLD und GalaxyDock. Im Vergleich zur leistungsstärksten Methode Rosetta GALigandDock schnitt PLACER im Bereich niedriger Präzision (Anteil der Komplexe mit Liganden-RMSD < 2 Å) besser ab (82,41 TP3T vs. 73,61 TP3T), im Bereich hoher Präzision (RMSD < 1 Å) jedoch etwas schlechter (41,81 TP3T vs. 51,61 TP3T).
Die Leistung von PLACER ist dennoch bemerkenswert, da es im Gegensatz zu anderen Methoden nicht speziell für das Docking von nicht-natürlichen Protein-Kleinmolekül-Paaren trainiert wurde. PLACER kann die Konformation von Kleinmolekülen und Proteinseitenketten von Grund auf rekonstruieren, während andere Testmethoden hauptsächlich auf den Koordinaten des Eingabeproteins basieren.
Design des aktiven Zentrums eines Enzyms
Die Anwendung von PLACER im Retro-Aldolase-Design ist besonders bemerkenswert. Das Forschungsteam führte 50 wiederholte Simulationen der RA95-Serie von Retro-Aldolasen und ihrer weiterentwickelten, verbesserten Versionen durch und analysierte die Konformationsvielfalt des aktiven Lysins und seines kovalenten Zwischenprodukts. Die Ergebnisse zeigten, dass PLACER für Enzyme mit geringer Aktivität im ursprünglichen Computerdesign hochdiverse Konformationssätze generierte, was auf einen Mangel an Präorganisation hindeutet; bei den aktiveren, weiterentwickelten Versionen hingegen wurden die Konformationssätze zunehmend geordneter. Dies legt nahe, dass…Das Fehlen einer Vororganisation ist ein großes Manko im frühen Stadium der Enzymentwicklung, während PLACER ein schnelles Bewertungsinstrument bietet, das zur Steuerung der Enzymentwicklungsarbeit eingesetzt werden kann.
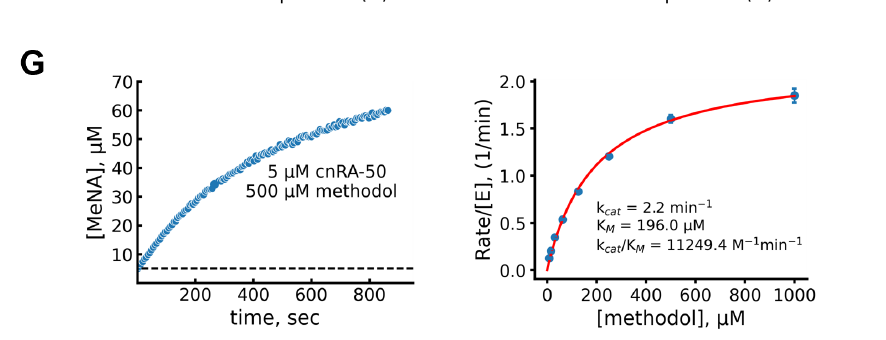
Darüber hinaus entwickelte das Team ein neuartiges Aldosteron-Reversionsenzym basierend auf einer NTF2-ähnlichen Faltung und untersuchte mithilfe von PLACER den Zusammenhang zwischen dem Grad der Präorganisation und dem kcat/KM-Wert. Die Ergebnisse zeigten, dass PLACER für hochgradig präorganisierte Designs im Allgemeinen eine höhere katalytische Effizienz vorhersagte. Das aktivste Design, cnRA-50, erreichte einen kcat/KM-Wert von 11.000 M⁻¹min⁻¹, der signifikant höher war als bei früheren computergestützten Designs und sich der Aktivität von Designs annäherte, die mit den neuesten RFdiffusion- und ProteinMPNN-Methoden erstellt wurden.
Das Forschungsteam prognostiziert,PLACER-basierte Konformationssatzgenerierungsmethoden werden in großem Umfang für die Strukturmodellierung komplexer Nicht-Protein-Moleküle in isolierten Zuständen oder Proteinumgebungen sowie für die Bewertung von Enzymdesign und Protein-Kleinmolekül-Konjugatdesign eingesetzt werden.
Professor David Baker: Ein Pionier, der sich seit langem mit computergestütztem Proteindesign beschäftigt.
Am 9. Oktober 2024 wurde dem renommierten Pionier des Proteindesigns, Professor David Baker, zusammen mit den AlphaFold2-Entwicklern Demis Hassabis und John M. Jumper von DeepMind der Nobelpreis für Chemie 2024 verliehen.
Professor David Baker beschäftigt sich seit Langem mit computergestütztem Proteindesign und stellt Open-Source-Tools für Deep Learning wie RoseTTAFold, RFdiffusion und ProteinMPNN zur Verfügung, um die Entwicklung neuartiger Proteine zu ermöglichen. Durch die Gründung eines Unternehmens hat er zudem die Industrialisierung dieser Technologien vorangetrieben und sich damit zu einem weltweit anerkannten Experten auf diesem Gebiet entwickelt. In seinen jüngsten Forschungsarbeiten hat sein Team bedeutende Durchbrüche in vielen neuen Bereichen erzielt.
Bei der Entwicklung neuer Medikamente nutzen Forscher beispielsweise häufig Proteine als zentrale Zielstrukturen, indem sie den Wirkstoff an strukturell stabile Proteine binden, um in den Krankheitsverlauf einzugreifen. Die gezielte Behandlung natürlich vorkommender, ungeordneter Proteine (IDPs), denen eine klar definierte Struktur, Sequenz und Konformationspräferenzen fehlen, stellt jedoch weiterhin eine Herausforderung dar. Vor diesem Hintergrund schlug das Team um David Baker im August 2025 eine Proteindesignstrategie namens Logos vor, die auf einer induzierten Anpassungsstrategie basiert. Logos entwickelt Bindungsproteine, die sich an 39 verschiedene ungeordnete Aminosäuresequenzen anpassen können. Dadurch stehen mehr Proteine als Zielstrukturen für die Entwicklung neuer Medikamente zur Verfügung, was die Forschung in den Bereichen Krebs und Alzheimer potenziell beschleunigen kann.
Titel des Papiers:Design von Proteinen, die an intrinsisch ungeordnete Regionen binden
Papieradresse:https://www.science.org/doi/10.1126/science.adr8063
Am 18. September 2025 stellte das Team um David Baker das All-Atom-Diffusionsmodell RFdiffusion3 (RFD3) vor, das die Entwicklung von biomolekularen Interaktionen auf Atomebene ermöglicht. Dieses Modell kann Proteinstrukturen im Kontext von Liganden, Nukleinsäuren und anderen Nicht-Protein-Clustern generieren und ist einfacher und effizienter als bisherige Methoden. In einer Reihe von Computersimulations-Benchmarks übertraf RFdiffusion3 frühere Methoden bei einem Rechenaufwand von nur einem Zehntel.
Titel des Papiers:Neuentwicklung von biomolekularen Interaktionen auf Atomebene mit RFdiffusion3
Papieradresse:https://www.biorxiv.org/content/10.1101/2025.09.18.676967v1
Natürliche Ionenkanäle spielen eine entscheidende Rolle in biologischen Systemen, und ihre künstlich hergestellten Varianten finden breite Anwendung in chemogenetischen Werkzeugen und Sensoren. Während Proteindesign bereits zur Konstruktion von Transmembranproteinen mit porösen Strukturen genutzt wurde, stellte die Entwicklung „selektiver Filter“ – also solcher mit präzisen Aminosäureseitenketten, die spezifische Ionen binden – wie natürliche Ionenkanäle eine technologische Herausforderung dar. Im Oktober 2025 gelang es dem Team um David Baker erstmals, mithilfe künstlicher Intelligenz einen neuartigen Calciumionenkanal von Grund auf zu entwickeln. Diese Studie zeigt, dass selbst komplexe biochemische Funktionen, die wir nur teilweise verstehen, mithilfe von KI nun von Grund auf konstruiert werden können.
Titel des Papiers:Bottom-up-Design von Ca²⁺-Kanälen ausgehend von einer definierten Selektivitätsfiltergeometrie
Papieradresse:https://www.nature.com/articles/s41586-025-09646-z
Angesichts ihrer jüngsten Erfolge gestalten Professor David Baker und sein Team die Proteinforschung in einem atemberaubenden Tempo um – von der Logos-Strategie, die an natürlich ungeordnete Proteine binden kann, über RFdiffusion3, das die Gestaltung molekularer Interaktionen auf atomarer Ebene ermöglicht, bis hin zur bahnbrechenden Forschung zur erstmaligen De-novo-Konstruktion von Calciumionenkanälen. Bakers Team treibt die computergestützte Proteinentwicklung kontinuierlich von der Theorie in die Praxis voran. Ihre Arbeit erweitert nicht nur die Grenzen des biomolekularen Designs, sondern macht die Zukunft des „Aufbaus von Lebensfunktionen mithilfe von Algorithmen“ immer deutlicher.
Referenzlinks:
1.https://www.biorxiv.org/content/10.1101/2024.09.25.614868v2
2.https://www.thepaper.cn/newsDetail_forward_31663354
3.https://www.biorxiv.org/content/10.1101/2025.09.18.676967v1
4.https://www.nature.com/articles/s41586-025-09646-z








