Command Palette
Search for a command to run...
トロント大学などが提案したdnaHNetは、推論速度を3倍向上させ、ゲノム学習の計算コストを約4分の1に削減する。

ゲノムは生物のすべての遺伝情報を担っており、細胞機能、個体発生、そして種の進化の方向性を決定する。配列の中に隠された「DNA構文」は、生命を支配する根本的な法則を構成しており、現代生物学が早急に解決しなければならない核心的な問題の一つである。この文法を理解することは、基礎的な科学知識に関わるだけでなく、疾病診断、医薬品開発、合成生物学といった重要な応用分野の開発にも直接影響を与える。
近年、大規模な配列データで事前学習された基本モデルが、この問題を解決するための重要な手段となりつつある。計算能力、データ規模、モデルパラメータの継続的な向上に伴い、これらのモデルは「スケーリング則」に類似した性能向上傾向を示している。Nucleotide TransformerやEvoに代表されるモデルは、パラメータ規模を数十億にまで拡大し、異種配列で学習することで、変異効果予測や調節エレメント解析などのタスクにおいて著しい進歩を遂げている。
しかし、DNA配列は本質的に境界が不明瞭なヌクレオチドの連続鎖であり、これは自然言語とは根本的に異なる。現在使用されている2つの主要なモデリングパラダイムは、固定単語分割と単一ヌクレオチドレベルのモデリングは、それぞれ表現力と計算効率の間に明確なトレードオフ関係が存在する。前者は生物学的機能単位を損傷する可能性があり、後者は計算コストが高い。したがって、計算の実現可能性と生物学的忠実性のバランスをより良く取ることが重要な課題となっている。潜在的な解決策として動的単語分割が挙げられるが、体系的な検討が依然として必要である。
この文脈では、dnaHNetモデルは、トロント大学、カナダのベクター人工知能研究所、米国のアーク研究所などの機関によって共同で提案され、これは、前述のボトルネックを克服するための新しいアプローチを提供するものです。関連する研究成果は、「dnaHNet:ゲノム配列学習のためのスケーラブルで階層的な基盤モデル」と題され、arXivにプレプリントとして公開されています。
研究のハイライト
* dnaHNetの計算効率はStripedHyena2を上回り、推論速度はTransformerの3倍以上速い。
* 圧縮率スケジューリングやエンコーダー・デコーダーのバランス調整など、最適なトレーニング戦略を提案します。
* 変異効果予測や遺伝子必須性分類など、サンプル数ゼロのタスクにおいて最先端に到達した。
* 文脈依存的な生物学的単語分割を学習し、コドン、プロモーター、遺伝子間領域などの機能領域に適応することができます。

用紙のアドレス:
https://arxiv.org/abs/2602.10603
弊社の公式WeChatアカウントをフォローし、バックグラウンドで「dnaHNet」と返信すると、PDF全文を入手できます。
モデルのトレーニングと評価のための多段階ゲノムデータセットの設計
モデルのトレーニングとシステム評価を支援するため、本研究では多層データシステムを構築した。事前学習データは、ゲノム分類データベース(GTDB)の処理済みサブセットから取得されます。このプロセスは、EvoモデルのOpenGenomeデータセットに対するフィルタリング、品質管理、および冗長性除去の手順に厳密に従って実施されました。スクリーニング基準には、アセンブリの完全性、汚染レベル、マーカー遺伝子含有量などの主要な指標が含まれており、スクリーニング後、各種レベルのクラスターごとに1つの代表的なゲノムのみが保持されました。
最終的なデータセットは85,205種の原核生物を対象とし、17,648,721個の配列を含んでいる。ヌクレオチドの総数は約1440億個である。すべての配列は完全なゲノムから抽出され、最大8,192ヌクレオチドの重複しないセグメントに分割された。
評価の観点から、研究者らはモデルの能力を包括的に検証するために、3つの補完的な次元からテストセットを構築した。まず、ローカルコーディングの適合性のレベルではMaveDBに登録されている大腸菌K12由来の、合計21,250データポイントからなる12個のヌクレオチドレベルの実験データセットを用いて、局所的なコーディング文法とタンパク質の適応度ランドスケープを特徴付けるモデルの能力を評価した。
第二に、全ゲノム規模での機能評価の観点からEssential Gene Database (DEG) に基づいて、62種の細菌種に対してバイナリ必須性タグが構築されました。関連する配列と注釈はNCBIから取得され、DEGエントリー名との配列同一性が99%より大きいものが必須遺伝子のラベル付け基準として使用されました。これにより185,226個のデータポイントが得られ、これらを用いて、モデルが長距離依存性とゲノムコンテキストを統合する能力を評価しました。
やっと、構造的解釈可能性の観点から、枯草菌ゲノムを例にとると、その機能アノテーションを組み合わせることで、配列を異なる機能領域に分割する。モデル分割結果と実際の生物学的構造とのアライメントを分析することで、構造モデリング能力を検証する。
dnaHNetモデル:単語分割なしの自己回帰フロンティアモデル
dnaHNetは、明示的なセグメンテーションツールを必要としないゲノムベースのモデルです。鍵となるのは「動的セグメンテーション」メカニズムであり、これによりモデルはシーケンス内の構造単位を自律的に学習することができる。この設計は、固定された単語分割による生物学的機能セグメントの断片化を回避し、ヌクレオチドごとのモデリングに伴う計算負荷を軽減することで、表現力と計算効率のより良いバランスを実現します。
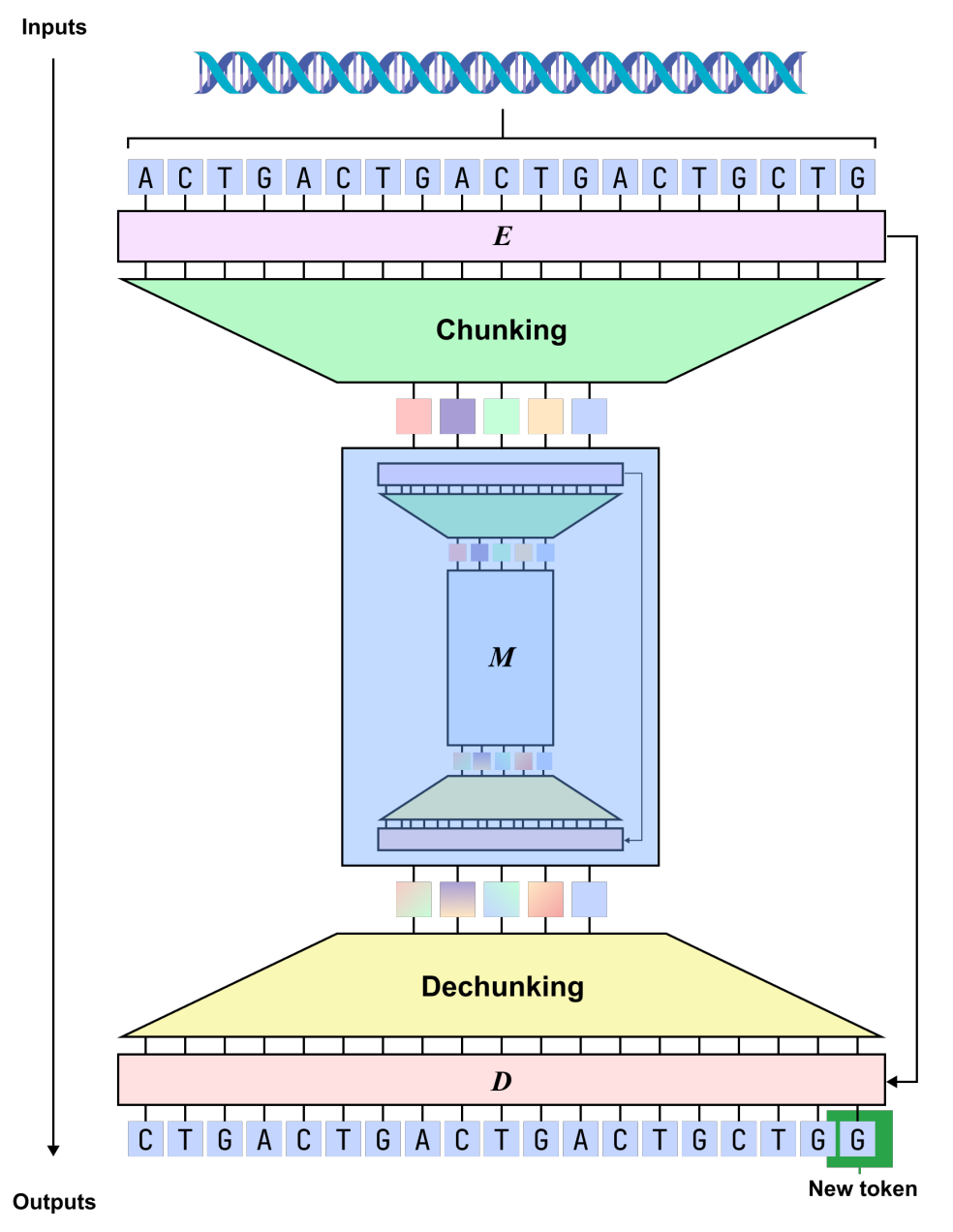
モデリング形式の観点から言えば、dnaHNetは、ゲノム学習を自己回帰的な配列予測タスクに統合し、既存のコンテキストに基づいて次のヌクレオチドを予測します。全体のアーキテクチャは階層構造を採用しており、各層はエンコーダ、バックボーンネットワーク、デコーダで構成されています。エンコーダはルーティングメカニズムを通じて、配列内で情報が大きく変化する場所(コドン境界や調節領域など)を特定し、それに応じて配列を暗黙的なブロック表現に圧縮します。バックボーンネットワークは、MambaとTransformerを組み合わせたハイブリッド構造を採用し、長距離依存性と重要な情報相互作用の両方を考慮して圧縮された配列をモデル化します。デコーダは、表現をヌクレオチド解像度にアップサンプリングし、予測結果を出力します。
この基盤の上に、dnaHNetはゲノムデータ向けにいくつかの重要な最適化を行いました。まず、パラメータ割り当てに関して、局所構造を特徴付ける能力を高めるために、エンコーダとデコーダに約301 TP3Tのモデル容量が割り当てられました。
第二に、2段階の層状圧縮設計を採用:第1段階では、コドンなどの小規模なパターンを捉えることに重点を置き、第2段階では、より広範囲の機能構造をモデル化することで、圧縮効率と情報忠実度のバランスを実現します。さらに、トレーニングプロセスには自己回帰予測損失と圧縮率の制約が組み込まれており、予測精度を維持しながら計算コストを効果的に制御できます。
推論段階では、モデルは境界確率に基づいてブロック分割方法を動的に決定するため、モデリングの粒度を状況に合わせて調整することができ、実際のゲノム構造により近いものとなる。
dnaHNetは計算コストを3.89倍削減し、他のマルチタスクアプリケーションよりも優れた性能を発揮します。
dnaHNetの性能を体系的に評価するために、本研究では、主流の2つの長鎖ゲノムモデルであるStripedHyena2とTransformer++と比較した。これらの実験は、スケーリング特性、ゼロサンプル変動効果の予測、遺伝子の必要性の予測、生物学的構造モデリングなど、複数の側面を網羅している。
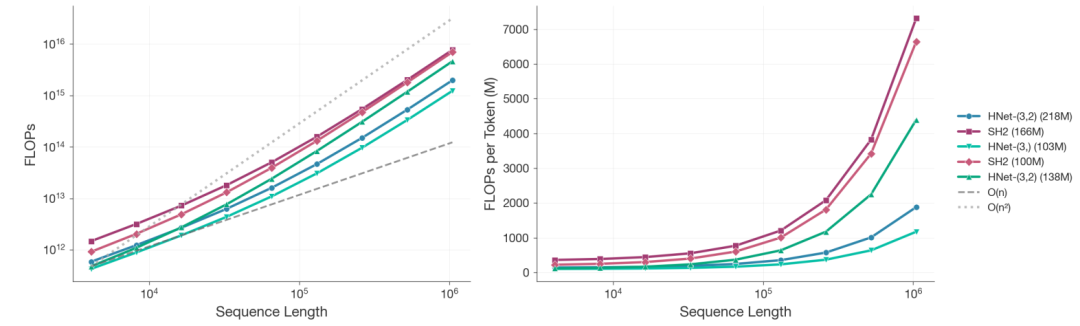
スケーリング分析では、研究者たちは、一定の計算予算の下で、さまざまなサイズの 100 を超えるモデルをトレーニングしました。配列の長さが 10⁶ ヌクレオチドに達し、総計算能力が 8 × 10¹⁹ FLOPs になったとき、2億1800万個のパラメータを持つdnaHNetの計算コストは、1億6600万個のパラメータを持つStripedHyena2と比較して約3.89倍削減される。2段階構造は、1段階構造よりもさらに効率的である。
パープレキシティに基づくべき乗則フィッティングの結果は、StripedHyena2が同等の性能レベルを達成するには、dnaHNetの約3.75倍の計算能力が必要となる。さらに、dnaHNetの最適なデータパラメータ構成は、従来のスケーリング法則から大きく逸脱しています。同じ計算能力の下で、dnaHNetのトレーニングトークン数は1400億に達するのに対し、比較モデルはわずか680億で、まだ収束していません。
下流タスクでは、dnaHNetは、サンプル数ゼロのタンパク質変動効果予測(MaveDB)と遺伝子必要性予測(DEG)の両方において、比較対象モデルを常に上回る性能を発揮します。さらに、その利点は計算能力の向上に伴ってさらに拡大する。これは、その動的なブロックベースのメカニズムと階層的なアーキテクチャが、局所的なコーディング構文と全体的なコンテキスト情報をより効果的に統合できることを示しており、それによって生物学的機能を特徴付ける能力が向上する。
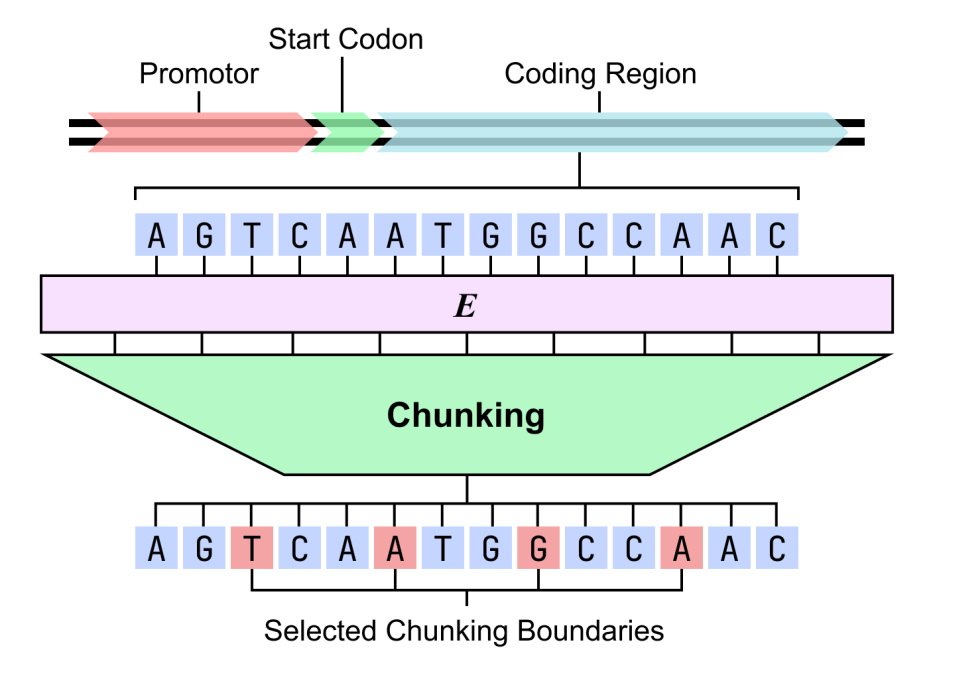
構造的解釈可能性に関して、枯草菌ゲノムを2段階のdnaHNetモデルを用いて解析した。その結果、このモデルは生物学的に意味のある階層構造を自発的に学習できることが示された。第1段階ではコドンに対する感度が高く、コーディング領域のトリプレットパターンを正確に捉えることができた。第2段階では機能構造に重点が置かれ、プロモーター、開始コドン、および遺伝子間領域はコーディング領域よりも有意に高いセグメンテーション確率を示した。
この結果は、このモデルは、高性能な予測能力を備えているだけでなく、教師なし学習条件下でゲノムの機能的組織を再構築することもできる。これは、「DNA文法」を解析するための、解釈可能な計算経路を提供する。
結論
全体として、dnaHNetは配列分割方法を事前に定義するのではなく、モデルがそれらを自動的に学習できるようにしました。実験では、この動的で階層的なモデリングが計算効率を向上させるだけでなく、ゲノムのマルチスケール構造をより適切に反映することが実証されています。長期的には、意味のある生物学的単位を安定的に学習できれば、ゲノム内の定式化が困難なパターンを明らかにし、変異予測、機能発見、合成設計における新たな研究の道を開く可能性を秘めています。