Command Palette
Search for a command to run...
En Concevant Des Substrats Hautement Sélectifs, Le MIT Et Harvard Collaborent Pour Découvrir De Nouveaux Modèles De Clivage Des Protéases Grâce À l'IA générative.

Au sein du réseau complexe de réactions biochimiques des organismes vivants, les protéases clivent spécifiquement les liaisons peptidiques, régulant ainsi avec précision une série de processus vitaux essentiels, de la coagulation sanguine et la réparation tissulaire aux réponses immunitaires et même à la progression du cancer. Un dysfonctionnement de ces protéases entraîne souvent directement l'apparition et le développement de diverses maladies graves. Par conséquent, l'élucidation des mécanismes d'action des protéases et la régulation précise de leur activité constituent non seulement un enjeu fondamental en sciences de la vie, mais aussi une avancée cruciale pour le développement de nouvelles méthodes diagnostiques et thérapeutiques.
La clé pour atteindre cet objectif,La clé réside dans la recherche de substrats peptidiques hautement « adaptés ».Ils peuvent être utilisés comme sondes moléculaires pour suivre l'activité enzymatique, conçus comme inhibiteurs pour bloquer une activité anormale, ou même servir d'« interrupteurs d'activation conditionnelle » dans les systèmes d'administration de médicaments pour réaliser une thérapie ciblée.
Cependant, concevoir des substrats peptidiques à la fois rapidement clivés par les protéases cibles et hautement sélectifs (reconnus uniquement par cette enzyme, évitant ainsi les réactions croisées avec d'autres protéases) a toujours constitué un défi majeur pour la communauté scientifique. Ce problème découle des interactions biochimiques complexes entre les protéases et leurs substrats : pour s'adapter à diverses fonctions physiologiques, les protéases ont développé une large spécificité de clivage, et leurs sites actifs doivent se lier précisément aux substrats peptidiques (généralement d'une dizaine d'acides aminés). Même en considérant uniquement des peptides synthétiques de 10 acides aminés, à partir des 20 acides aminés naturels courants, le nombre de combinaisons de séquences théoriques peut atteindre environ 20¹⁰ (près de 10¹³), créant un espace d'exploration quasi infini. Et ce n'est pas tout…Les protéases aux fonctions similaires proviennent souvent d'un ancêtre commun et possèdent des structures de site actif similaires, ce qui les rend très susceptibles à la « reconnaissance croisée ».Cela rend particulièrement difficile la sélection de substrats très spécifiques parmi un grand nombre de possibilités.
Pour surmonter cet obstacle, les chercheurs ont entrepris de nombreuses tentatives. Les méthodes traditionnelles s'appuient souvent sur des sites de clivage connus ou sur des informations enzymatiques de protéines naturelles, ce qui entraîne une faible efficacité et la difficulté d'obtenir des substrats artificiels idéaux. La conception rationnelle basée sur les connaissances en biologie chimique est généralement complexe, présente un débit limité et cible principalement des protéases uniques, ce qui rend son passage à l'échelle industrielle difficile. Ces dernières années, bien que les technologies de criblage à haut débit aient amélioré l'efficacité dans une certaine mesure, elles souffrent encore de limitations telles qu'une mise en œuvre complexe et un coût élevé.La plupart des méthodes de prédiction informatique existantes ne peuvent que déterminer « s'il faut couper » et ne peuvent pas trier avec précision l'efficacité de la coupe, ne répondant ainsi pas aux besoins de la recherche approfondie sur les mécanismes et des applications d'ingénierie.
Dans ce contexte,Le MIT et l'Université Harvard ont proposé conjointement CleaveNet, un flux de conception de bout en bout basé sur l'intelligence artificielle.En faisant fonctionner en synergie les modèles prédictifs et génératifs, cette approche vise à révolutionner le paradigme existant de la conception des substrats de protéases et à fournir des solutions entièrement nouvelles pour la recherche fondamentale et le développement biomédical connexes.

Adresse du document :
https://www.nature.com/articles/s41467-025-67226-1
Suivez notre compte WeChat officiel et répondez « CleaveNet » en arrière-plan pour obtenir le PDF complet.
Autres articles sur les frontières de l'IA :
Validation inter-scénarios qui renforce la capacité de généralisation du modèle CleaveNet, à l'aide d'ensembles de données provenant de multiples scénarios expérimentaux.
Pour développer et valider le modèle CleaveNet, cette étude a intégré deux ensembles de données qui différaient significativement en termes de composition de séquences et de méthodes expérimentales afin de garantir la fiabilité et la capacité de généralisation du modèle.
L'ensemble de données principal utilisé par les chercheurs provient d'une étude publiée qui a systématiquement caractérisé l'activité de clivage d'une bibliothèque de substrats contenant environ 18 500 décapeptides synthétiques contre 18 métalloprotéinases matricielles (MMP) à l'aide de la technologie d'affichage d'ARNm.Chaque combinaison substrat-protéase correspond à un score d'efficacité de clivage standardisé (Zₛₘ) pour quantifier l'intensité relative du clivage.
Afin de garantir davantage la rigueur de l'évaluation et d'éviter toute surestimation due à la similarité des séquences,Les chercheurs ont effectué un filtrage par homologie sur l'ensemble de test initial :Les chercheurs ont calculé la distance de Levenstein minimale entre chaque séquence de test et toutes les séquences de l'ensemble d'entraînement, puis ont éliminé 816 séquences présentant une distance inférieure à 3 et une forte similarité avec l'ensemble d'entraînement. Ils ont ainsi obtenu un ensemble de test d'affichage d'ARNm contenant 2 901 séquences non chevauchantes. Ce sous-ensemble n'a été utilisé à aucune étape de l'entraînement du modèle et a servi exclusivement à la validation interne des performances.
Afin de tester indépendamment l'adaptabilité du modèle face à des contextes biochimiques radicalement différents,L'étude a également introduit un ensemble de données hors distribution totalement indépendant, appelé « ensemble de test de fluorescence ».Cet ensemble de données contient 71 peptides synthétiques de longueurs variables (7 à 14 acides aminés), dont l'activité de clivage a été validée pour sept protéines MMP recombinantes par des expériences in vitro classiques basées sur le transfert d'énergie par résonance de fluorescence (FRET). Cet ensemble diffère fondamentalement de l'ensemble de données initial généré par la technologie d'affichage d'ARNm, notamment en termes de distribution de la longueur des peptides, de composition en acides aminés et, surtout, de principes de détection expérimentaux. Cette conception délibérée constitue un point de référence essentiel pour évaluer la capacité du modèle CleaveNet à s'affranchir de conditions expérimentales spécifiques et à identifier des motifs biochimiques universels.
CleaveNet prédit et génère des boucles de collaboration fermées.
Comme le montre la figure ci-dessous, le cœur de CleaveNet est constitué de deux modules de calcul complémentaires et collaboratifs : le module de prédiction (CleaveNet Predictor) et le module de génération (CleaveNet Generator).Ensemble, ils forment une boucle fermée complète « conception-évaluation ».
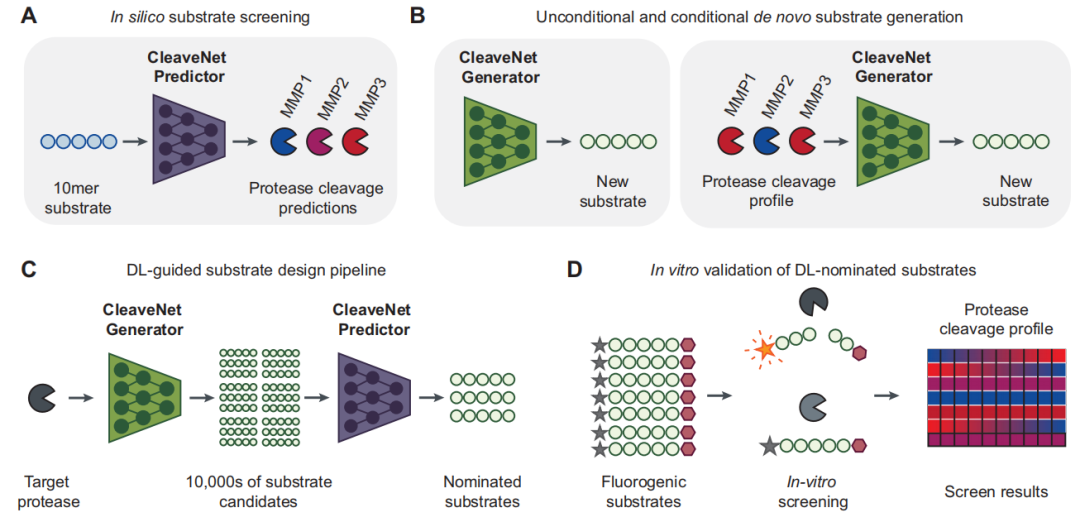
Le module de prédiction vise à résoudre le problème de l'évaluation rapide et précise de l'activité de clivage des substrats candidats à partir d'un espace de séquences massif.Les chercheurs l'ont conçu comme un modèle de régression de fonction de séquence à sorties multiples. Plus précisément, le modèle prend une séquence d'acides aminés en entrée et sa tâche principale consiste à fournir simultanément le score de coupure prédit (Ŵₛₘ) de la séquence pour les 18 MMP, et à estimer simultanément l'incertitude (σₛₘ) de chaque prédiction.
Pour obtenir une plus grande robustesse prédictive, cette étude a utilisé une stratégie d'ensemble de modèles :Cinq modèles de prédiction identiques ont été entraînés indépendamment sur l'ensemble d'entraînement d'affichage d'ARNm, le score de prédiction final correspondant à la moyenne de leurs sorties. L'incertitude des prédictions a été quantifiée par l'écart type de ces cinq résultats. De plus, grâce à la définition d'un seuil ajustable (Zₜ), le modèle peut facilement convertir les scores de prédiction continus en un jugement binaire (« coupé » ou « non coupé »), s'adaptant ainsi à différents scénarios de criblage.
Pour construire le modèle prédictif, cette étude a comparé systématiquement deux architectures courantes en modélisation de séquences : les réseaux LSTM bidirectionnels et les Transformers. Les premiers excellent dans la capture des dépendances entre séquences, tandis que les seconds, grâce à leur mécanisme d’attention, peuvent modéliser globalement les interactions entre acides aminés et constituent actuellement le choix privilégié pour la représentation du langage protéique. Compte tenu de son potentiel démontré sur des données plus vastes et plus diversifiées,Les chercheurs ont finalement choisi l'architecture Transformer comme base pour le prédicteur CleaveNet.
L'objectif du module de génération est de parvenir à une conception automatisée et intelligente des substrats candidats.Cette étude a entraîné un modèle génératif basé sur un Transformer autorégressif, lui permettant d'apprendre les préférences universelles de découpe MMP inhérentes à l'ensemble de données à partir de la représentation de l'ARNm.Ce modèle peut générer un grand nombre de séquences peptidiques nouvelles et plausibles sans aucune condition d'entrée supplémentaire.
Afin d’évaluer scientifiquement la valeur des modèles génératifs plutôt que de simplement reproduire le hasard, les chercheurs ont développé une méthode de base robuste appelée « contrôle indépendant du site ».Cette méthode ne prend en compte que la distribution indépendante de chaque position d'acide aminé dans les données d'entraînement, puis effectue un échantillonnage aléatoire sur cette base pour générer des séquences.En comparant les séquences générées par CleaveNet avec cette séquence de référence selon de multiples dimensions, nous pouvons clairement identifier les schémas biochimiques complexes appris par le modèle qui vont au-delà des simples associations statistiques.
L'étroite collaboration entre les modules de prédiction et de génération permet aux chercheurs de générer d'abord une bibliothèque de candidats diversifiée, puis d'effectuer un criblage virtuel efficace et précis sur celle-ci, fournissant ainsi un puissant moteur de calcul pour la vérification expérimentale ultérieure.
CleaveNet permet un contrôle sélectif et précis.
Après avoir achevé la construction du modèle, cette étude a mené une vérification expérimentale multiniveau et systématique des performances de CleaveNet, et les résultats ont pleinement démontré la valeur exceptionnelle de ce processus en termes de précision de prédiction, de rationalité de génération et d'efficacité d'application pratique.
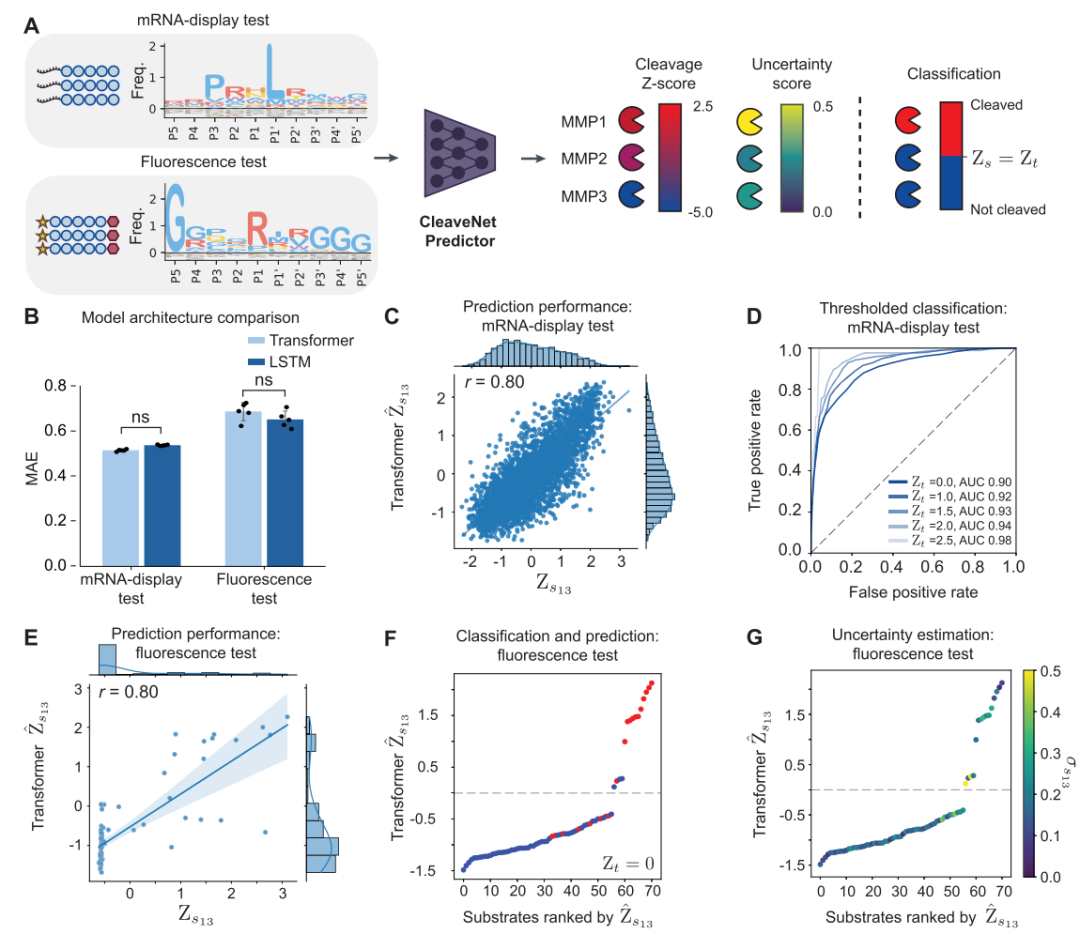
d'abord,CleaveNet Predictor démontre d'excellentes capacités de prédiction sur les ensembles de tests internes et externes.Sur un jeu de test de filtrage par homologie (jeu de test d'affichage d'ARNm) non utilisé lors de l'entraînement, le score prédit par le modèle (Ŵₛₘ) pour MMP13 a montré une forte corrélation avec le score Z standardisé mesuré expérimentalement (Zₛₘ) (coefficient de corrélation de Pearson r = 0,80). Ses performances sont restées tout aussi robustes lorsque les prédictions continues ont été converties en une classification binaire « coupé/non coupé » : en traçant des courbes ROC (Receiver Operating Characteristic) et en calculant l'aire sous la courbe (AUC), les chercheurs ont constaté que le modèle conservait un pouvoir discriminant élevé pour différents seuils de décision, notamment au seuil de coupure universellement accepté (Zₜ = 2,5), où l'AUC a atteint 0,98. Des tests plus rigoureux ont été réalisés sur des jeux de test de fluorescence totalement indépendants, avec des méthodes expérimentales très différentes.
Bien que la longueur de la séquence, la composition en acides aminés et le principe de détection de cet ensemble de données soient différents de ceux des données d'entraînement, le score de coupure prédit par le modèle maintient toujours une forte corrélation positive avec la valeur expérimentale (r = 0,80 pour MMP13) et peut distinguer avec précision les séquences « coupées » et « non coupées » vérifiées expérimentalement.Cela confirme fortement que CleaveNet Predictor peut non seulement mémoriser les modèles de données d'entraînement, mais aussi capturer les lois biochimiques universelles régissant le clivage du substrat par les protéases.Elle possède une forte capacité de généralisation.
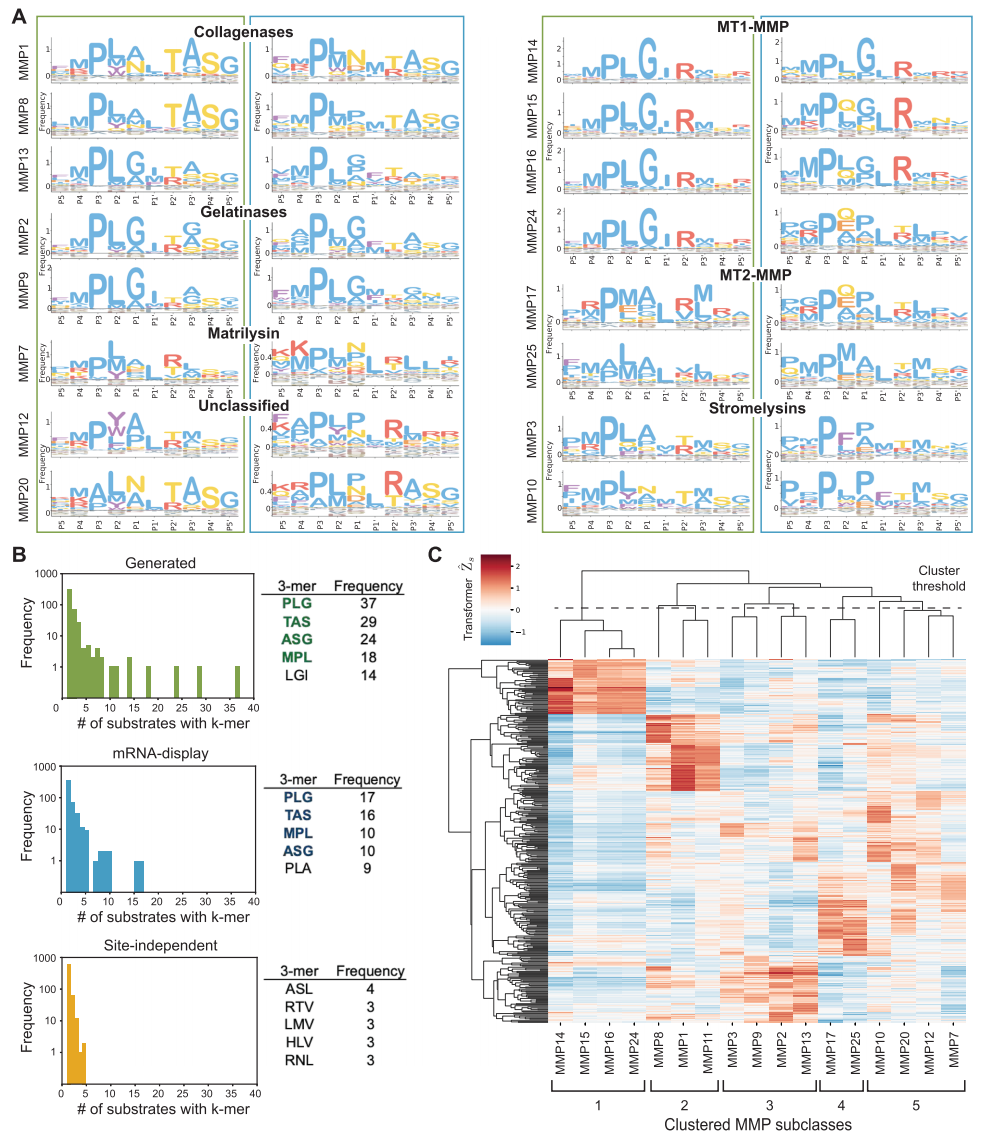
Deuxièmement,L'analyse bioinformatique des séquences générées par CleaveNet Generator par des chercheurs a révélé sa logique et sa nouveauté.Comparées aux séquences de « contrôle indépendant du site » basées uniquement sur un échantillonnage aléatoire des fréquences de position d'acides aminés individuels, les séquences générées par le modèle génératif reproduisent plus fidèlement les motifs de clivage classiques de la famille des MMP et présentent une distribution des acides aminés dans les régions clés de la poche de liaison au substrat qui ressemble davantage aux données expérimentales réelles. Plus important encore,Les séquences générées sont cohérentes avec l'ensemble de données réel en termes de propriétés biophysiques globales (telles que l'hydrophobicité et la charge).Cependant, une génération de haute qualité ne se limite pas à une simple réplication des données d'entraînement. L'analyse de la diversité des séquences a montré que la proportion de longs peptides synthétiques uniques partagés par les séquences générées et l'ensemble d'entraînement était extrêmement faible, indiquant que le modèle a évité le surapprentissage et a pu explorer de nouveaux espaces de séquences non couverts par les données d'entraînement.
Une analyse de regroupement fonctionnel plus poussée a révélé que les spectres d'activité de clivage prédits des substrats à score élevé générés par différentes MMP pouvaient être naturellement regroupés en fonction des relations phylogénétiques des domaines catalytiques des MMP.Ceci démontre que le modèle génératif n'apprend pas seulement les schémas de séquence apparents, mais capture aussi intrinsèquement des informations sur la différenciation fonctionnelle dans l'évolution des protéases.Ceci démontre la rationalité biologique des résultats obtenus.
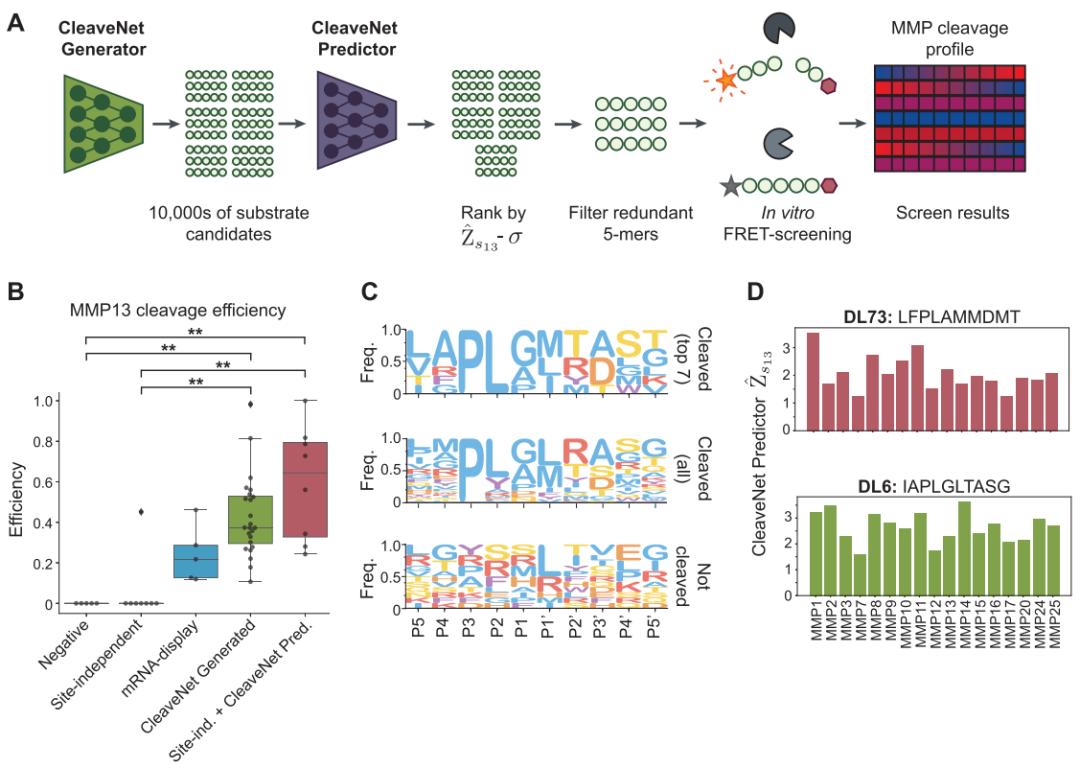
En définitive, la validité de tous les modèles informatiques a été confirmée par des expériences biochimiques in vitro. Les chercheurs ont synthétisé plusieurs ensembles de substrats candidats conçus par CleaveNet ciblant la MMP13, incluant des séquences générées directement par le modèle génératif et des séquences sélectionnées par le modèle prédictif. Les expériences de clivage par transfert d'énergie de résonance de fluorescence (FRET) ont donné des résultats convaincants.Les 24 substrats conçus à l'aide du pipeline CleaveNet ont tous été clivés avec succès par le MMP13 reconstruit, atteignant un taux de réussite de 100 % (TP3T).De plus, l'efficacité de coupe médiane était significativement supérieure à celle des substrats témoins positifs à haute efficacité connus dans l'ensemble d'entraînement. Ceci confirme la capacité de ce procédé à concevoir des substrats à haute efficacité.
Afin de démontrer le potentiel du procédé pour aborder des tâches plus complexes, telles que la conception de substrats hautement sélectifs, cette étude a mis en œuvre une stratégie de génération conditionnelle, en spécifiant une « sélectivité élevée pour la MMP13 » comme objectif dans le modèle génératif. Un criblage in vitro parallèle à grande échelle (95 paires de substrats pour 12 MMP différentes) a ensuite montré que les substrats générés par guidage conditionnel présentaient une sélectivité élevée.L'activité de clivage est significativement orientée vers la MMP13, ce qui entraîne une sélectivité plus élevée.
Il convient de souligner en particulier que certains des substrats conçus possèdent à la fois une efficacité de coupe élevée et une sélectivité élevée, une excellente combinaison extrêmement rare dans les données d'entraînement originales, ce qui met en évidence la puissante capacité de CleaveNet à explorer de nouveaux espaces de séquences de haute qualité.
En résumé, de la prédiction informatique précise à la génération de séquences pertinentes, puis à la vérification expérimentale concluante, une série de résultats interdépendants démontre que CleaveNet a construit une plateforme de conception de substrats de protéases efficace, fiable et performante. Cette recherche apporte non seulement une solution d'IA novatrice au défi classique de la régulation de l'activité des protéases, mais jette également les bases méthodologiques d'une nouvelle étude sur la fonction des protéases et le développement de médicaments associés.
Innovation pilotée par l'IA dans la conception de substrats de protéases
La technologie de conception de substrats de protéases assistée par l'IA de CleaveNet stimule l'innovation dans les domaines des sciences de la vie et de la biomédecine à l'échelle mondiale.
L'équipe de David Baker à l'Université de Washington a publié des recherches novatrices dans la revue Science.Pour la première fois, l'IA a été utilisée pour concevoir de toutes pièces une sérine hydrolase dotée d'un site actif complexe — l'une des plus grandes familles d'enzymes connues.L'étude a introduit un nouveau réseau d'apprentissage automatique, PLACER, qui a non seulement conçu avec succès une enzyme active capable de catalyser efficacement l'hydrolyse des esters, mais a également découvert de manière inattendue cinq nouveaux modèles de repliement des protéines, élargissant considérablement la diversité structurelle de cette famille d'enzymes.
* Titre de l'article : Conception informatique des hydrolases à sérine
* Lien vers l'article :
https://www.science.org/doi/10.1126/science.adu2454
Par ailleurs, une équipe conjointe de plusieurs universités européennes a développé un modèle général, basé sur l'architecture Transformer, capable de prédire avec précision les interactions protéase-substrat. Ce modèle intègre des données de clivage protéasiques issues de sources multiples à l'échelle mondiale, permettant ainsi une prédiction efficace des séquences de substrats chez différentes espèces. Sa capacité de généralisation a été validée par des recherches sur les protéases de divers pathogènes, notamment des bactéries et des virus, fournissant ainsi une base importante pour la conception de séquences en vue du développement de médicaments anti-infectieux.
Il est prévisible qu'avec la convergence continue de la biologie computationnelle, de l'intelligence artificielle et de la biologie synthétique, la conception des substrats de protéases évoluera d'une science alliant art et expérience vers un domaine de recherche hautement rationalisé et technique. Ceci accélérera non seulement le développement de nouveaux médicaments, d'outils de diagnostic et de biocatalyseurs écologiques, mais nous permettra aussi potentiellement de décrypter la logique sous-jacente de la régulation du vivant, inaugurant ainsi une nouvelle ère de programmation à la demande des fonctions vitales.








