Command Palette
Search for a command to run...
NVIDIA, Le MIT, l'Université d'Oxford, l'Université De Copenhague, Peptone Et d'autres Encore Publient Des Modèles Génératifs Et De Nouveaux Points De Référence Afin De Redéfinir Le Pouvoir Prédictif Des Assemblages De Protéines désordonnés.

Dans l'histoire de la biologie structurale, le principe selon lequel « la structure détermine la fonction » a longtemps été considéré comme une loi fondamentale quasi inébranlable. La conformation hélicoïdale classique de l'insuline et la structure tétramérique de l'hémoglobine ont toutes deux renforcé le consensus selon lequel les protéines doivent posséder une structure tridimensionnelle stable pour exercer leurs fonctions biologiques.
Cependant,La découverte des protéines intrinsèquement désordonnées (IDP) et de leurs régions intrinsèquement désordonnées (IDR)Cette conception traditionnelle est constamment remise en question. Ces cellules ne forment pas de structures fixes dans des conditions physiologiques, mais elles sont profondément impliquées dans des processus fondamentaux tels que la transduction du signal et la régulation de la transcription génique, et sont étroitement liées à des maladies humaines majeures comme le cancer et les maladies neurodégénératives.
Des recherches en biologie computationnelle ont par ailleurs révélé qu'environ 301 résidus d'acides aminés TP3T du protéome eucaryote se trouvent dans un état désordonné. Cela signifie que le désordre n'est pas « anormal », mais plutôt une composante normale des systèmes vivants. Cependant,La nature hautement dynamique des protéines désordonnées les rend difficiles à capturer de manière stable à l'aide des techniques expérimentales traditionnelles, et également difficiles à simuler avec précision de leur distribution conformationnelle à l'aide des méthodes de calcul conventionnelles.Cela constitue depuis longtemps un goulot d'étranglement technologique dans ce domaine.
Pour relever ce défi, une équipe conjointe composée de Peptone, une société britannique de développement de technologies d'analyse des protéines, de l'Université de Copenhague, de NVIDIA, de l'Université d'Oxford, du MIT, de l'Université Duke et d'autres, a proposé deux avancées majeures.L'un d'eux est le cadre d'évaluation du système PeptoneBench.Ce cadre intègre des données expérimentales multi-sources telles que SAXS, RMN, RDC et PRE, et combine des méthodes statistiques telles que la pondération par entropie maximale pour parvenir à une comparaison quantitative rigoureuse entre les observations expérimentales et les prédictions théoriques.Le second est le modèle génératif PepTron.L'entraînement sur un ensemble de données IDR synthétiques élargi améliore spécifiquement la capacité à modéliser les régions désordonnées, lui permettant de mieux capturer la diversité conformationnelle des protéines désordonnées.
L'équipe de recherche a utilisé PeptoneBench pour comparer systématiquement PepTron aux principaux outils de prédiction tels qu'AlphaFold2, Boltz2 et BioEmu. Les résultats ont montré que PepTron présentait une excellente concordance avec les résultats expérimentaux pour la prédiction des régions ordonnées et désordonnées, atteignant ainsi des performances de pointe. Grâce à ces avancées, un cadre plus précis et biologiquement réaliste pour la prédiction des structures protéiques à l'aide d'un « ensemble de conformations » est en train d'émerger, améliorant considérablement notre compréhension globale des protéines sur l'ensemble de leur spectre ordre-désordre.
Les résultats de recherche associés, intitulés « Amélioration des prédictions d’ensembles de protéines à travers le continuum ordre-désordre », ont été publiés en tant que prépublication sur bioRxiv.

Adresse du document :
https://www.biorxiv.org/content/10.1101/2025.10.18.680935v1
Suivez notre compte WeChat officiel et répondez « PepTron » en arrière-plan pour obtenir le PDF complet.
Autres articles sur les frontières de l'IA :
https://hyper.ai/papers
Construction systématique de PeptoneBench et d'ensembles de données expérimentales multi-sources
Les bases de données de protéines (PDB) sont les ressources publiques les plus fondamentales et les plus importantes en biologie structurale, mais il existe des lacunes structurelles importantes dans leur couverture des protéines intrinsèquement désordonnées (IDP) et de leurs régions désordonnées (IDR).Seules 31 entrées TP3T environ ont été marquées comme non ordonnées.Dans le protéome humain, cependant, la proportion de ces régions désordonnées atteint 20–30%.
Comme le montre la figure ci-dessous, ce biais systématique fait que la plupart des modèles de prédiction structurale « privilégient » naturellement les conformations stables, ce qui limite leur capacité à apprendre à partir d'états dynamiques et désordonnés sur le long terme. Pour compenser cette déficience,Les chercheurs ont introduit des bases de données supplémentaires telles que IDRome, qui contiennent une proportion non ordonnée d'environ 771 TP3T.Elle peut compléter la PDB en termes de distribution statistique. Cependant, cette base de données manque de données structurelles analysées dans le cadre d'expériences réelles, ce qui rend difficile son utilisation comme référence directe pour la modélisation et l'évaluation, et sa valeur applicative reste donc considérablement limitée.
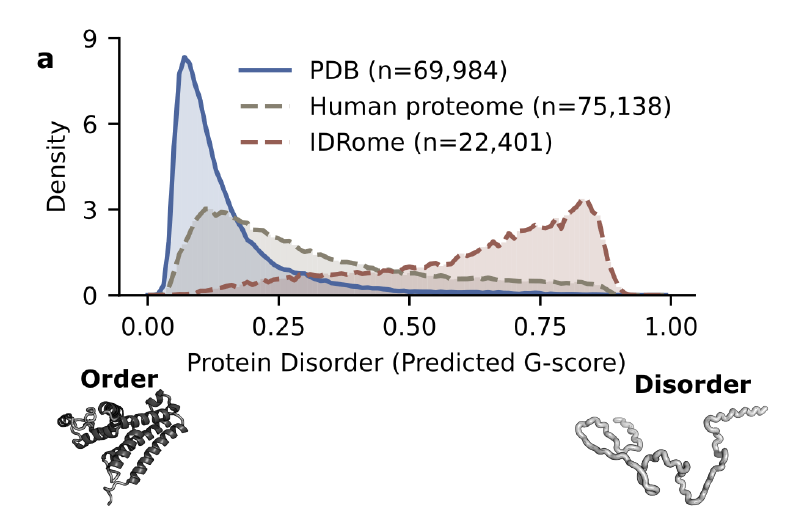
Pour surmonter les goulots d'étranglement des données susmentionnésLa première étape consiste à établir des indicateurs de désordre quantifiables et comparables.Cette étude utilise le score G moyen des protéines comme métrique principale, avec des valeurs allant de 0 (totalement ordonné) à 1 (totalement désordonné). Calculé à partir des données de déplacement chimique secondaire (DC) en RMN, il reflète avec précision la tendance à la formation de structures secondaires locales. Pour les protéines ne disposant pas de données DC expérimentales, l'équipe de recherche a utilisé le modèle d'apprentissage automatique ADOPT2, entraîné sur TriZOD, pour prédire le score G, obtenant ainsi une quantification unifiée de l'ensemble du spectre ordre-désordre.
S’appuyant sur ce constat, l’équipe a également souligné que le fait de se fier uniquement aux données structurelles des PDB ne permet pas d’évaluer objectivement la qualité des ensembles de conformations.Il est donc nécessaire de construire un ensemble de données expérimentales qui couvre l'ensemble de la plage ordonnée-non ordonnée.
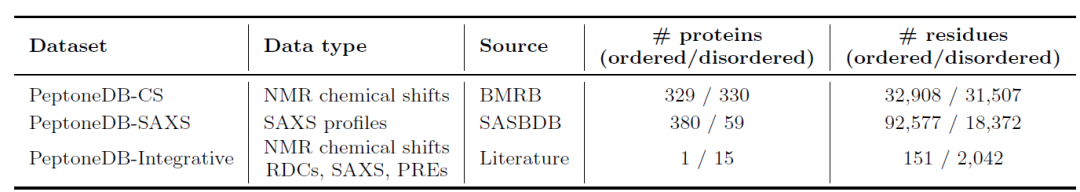
À cette fin, comme le montre le tableau ci-dessous, les chercheurs ont établi trois ressources de données complémentaires : PeptoneDB-CS (déplacements chimiques RMN issus de BMRB), PeptoneDB-SAXS (spectres SAXS issus de SASBDB) et PeptoneDB-Integrative (un ensemble de données dédié aux protéines intrinsèquement désordonnées intégrant de multiples données expérimentales orthogonales). Ces trois types de données présentent des structures différentes et des informations complémentaires : CS révèle les structures locales, SAXS reflète la conformation globale et Integrative permet la validation croisée.
D’après ces données, comme le montre la figure ci-dessous.Les chercheurs ont développé le cadre d'évaluation PeptoneBench pour quantifier la cohérence entre l'ensemble des conformations prédites et les données expérimentales.Le processus complet comprend : la standardisation et le prétraitement des ensembles de conformations ; la mise en correspondance de la structure prédite avec des observations comparables aux expériences à l’aide d’un modèle direct ; l’évaluation de la cohérence basée sur l’erreur quadratique moyenne normalisée (RMSE), intégrant les incertitudes du modèle et des expériences tout au long du processus. Les résultats finaux sont présentés sous forme de graphique RMSE-G, et les erreurs sont estimées par lissage de Lowes et rééchantillonnage bootstrap, puis synthétisées en un score agrégé PeptoneBench, constituant ainsi un standard quantitatif pour la comparaison directe des performances de différents outils.
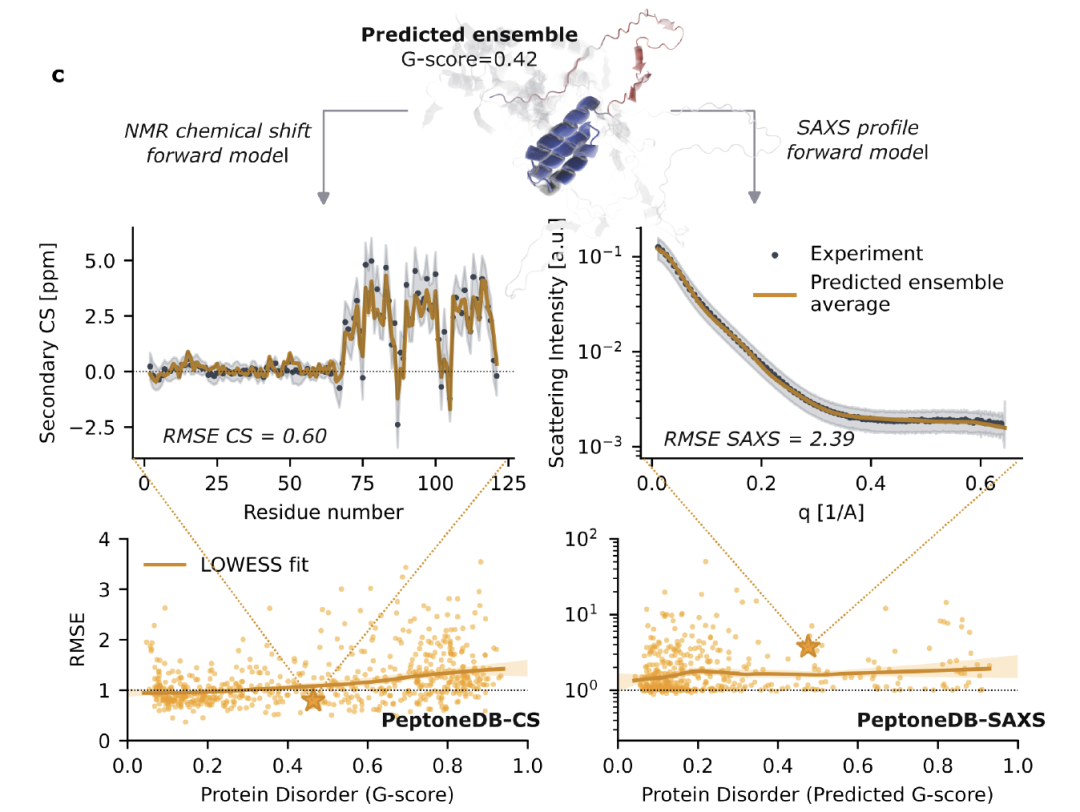
Il convient de souligner que certains ensembles initiaux de conformations présentant une erreur quadratique moyenne (RMSE) élevée peuvent en réalité être plus proches de la distribution expérimentale après pondération à l'aide de l'entropie maximale. Afin d'éviter de confondre des « poids incorrects » avec des « conformations manquantes »,PeptoneBench indique également l'erreur quadratique moyenne (RMSE) avant et après la pondération afin de distinguer entre le biais d'échantillonnage corrigible et la perte conformationnelle irrécupérable.Cette stratégie est particulièrement cruciale pour les IDP, qui sont très dynamiques et extrêmement sensibles aux conditions expérimentales : tant que le modèle génératif peut couvrir un espace conformationnel suffisamment riche, il peut s'adapter rapidement grâce au processus de repondération même si l'environnement expérimental est différent, améliorant ainsi considérablement la praticité et la fiabilité des résultats de prédiction.
PepTron : Un modèle conformationnel qui équilibre les protéines ordonnées et désordonnées
Le modèle PepTron proposé est un générateur de conformations protéiques basé sur l'architecture de correspondance de flux ESMFlow. Son objectif est de couvrir l'ensemble du spectre conformationnel, des conformations totalement ordonnées aux conformations hautement désordonnées, en générant un ensemble de conformations à la fois physiquement plausibles et structurellement diversifiées.
En termes d'architecture du modèle,PepTron est basé sur ESMFlow et implémenté dans le framework NVIDIA BioNeMo pour améliorer l'efficacité de l'entraînement et de l'inférence.Le modèle intègre le mécanisme d'attention triangulaire cuEquivariance et prend en charge la mise en correspondance des flux grâce au sous-package Modular Co-Design de BioNeMo. Le processus d'entraînement suit les bonnes pratiques de BioNeMo en matière de calcul distribué, combinant plusieurs stratégies parallèles et le calcul en précision mixte, ce qui permet une mise à l'échelle stable et efficace dans les environnements multi-GPU.
Il convient de souligner que PepTron ne repose ni sur l'alignement de séquences multiples (MSA), ni sur des pondérations ESM externes lors de la phase d'inférence. Il peut générer un ensemble complet de conformations avec un seul point de contrôle, ce qui simplifie considérablement le seuil d'utilisation.
Pour pallier le manque de données expérimentales sur la structure des régions désordonnées, l'équipe de recherche a construit un ensemble de données de structures synthétiques, IDRome-o, basé sur IDRome.Ils ont développé IDP-o, un outil de génération de structures protéiques basé sur l'assemblage de fragments, capable de générer à grande échelle des ensembles de conformations IDP physiquement plausibles à un coût extrêmement faible. IDP-o combine des stratégies d'assemblage de fragments et de croissance de chaînes hiérarchiques pour extraire des fragments de six résidus de la base de données AlphaFold, qui contient 214 millions de structures, capturant ainsi plus précisément les structures hélicoïdales transitoires dans les protéines désordonnées.
Il convient de noter que l'objectif d'IDR-o n'est pas de simuler une distribution d'équilibre particulière, mais de couvrir toutes les conformations raisonnables que la séquence peut adopter. Par conséquent, ses résultats sont particulièrement adaptés à un réajustement ultérieur par pondération d'entropie maximale et peuvent également servir de bibliothèque de conformations initiales de haute qualité pour les simulations de dynamique moléculaire.
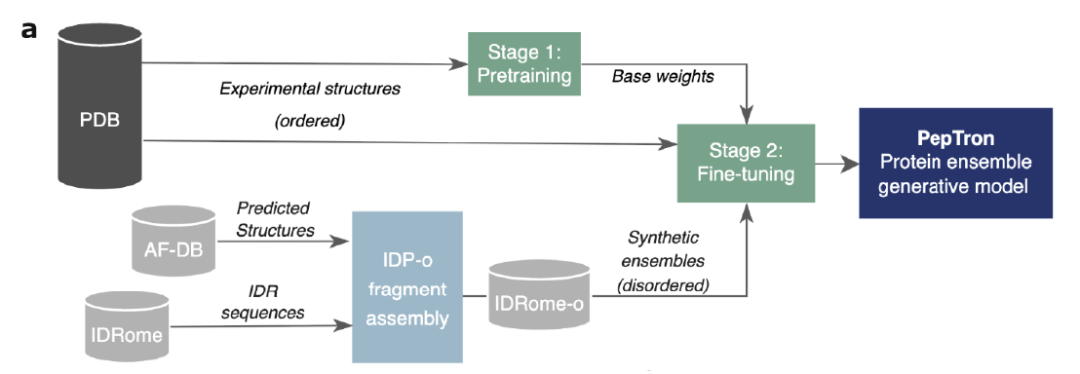
Pour surmonter le biais des modèles traditionnels qui tendent à prédire des structures stables, comme le montre la figure ci-dessous,PepTron utilise une stratégie d'entraînement hybride combinant données expérimentales et données synthétiques.Dans un premier temps, le modèle est pré-entraîné à l'aide des structures résolues expérimentalement issues de la base de données PDB. Ensuite, un ensemble de protéines désordonnées générées synthétiquement est introduit pour un ajustement fin, permettant ainsi au modèle d'apprendre pleinement la distribution continue des conformations ordonnées et désordonnées. Même avec des ressources de calcul limitées, cette stratégie améliore significativement les performances prédictives du modèle sur diverses protéines.
En ce qui concerne les procédures de formation spécifiques,La recherche a été divisée en deux phases :Dans une première étape, à partir des poids ESMFold, le module de correspondance de flux est réentraîné à l'aide des données PDB, et la plage de réduction de la longueur des séquences est étendue à 512 résidus. Lors de l'étape d'ajustement fin hybride, un jeu de données hybride, composé de structures expérimentales PDB et de données synthétiques IDRome-o, sert de données d'entraînement pour l'optimisation finale du modèle. Cette conception permet à PepTron d'explorer l'ensemble du spectre ordonné-désordonné, offrant ainsi une modélisation plus complète et réaliste de l'espace conformationnel dynamique des protéines.
Validation de modèles pour les conformations à spectre complet : une comparaison systématique des méthodes PepTron et Mainstream
L'équipe de recherche a ensuite utilisé le framework PeptoneBench pour évaluer systématiquement les performances de PepTron sur des données expérimentales totalement indépendantes de l'ensemble d'entraînement, et les a comparées à celles de modèles de référence tels que ESMFold, ESMFlow, AlphaFold2, Boltz2 et BioEmu. Parallèlement, elle a mené des tests spécifiques sur l'ensemble de données PeptoneDB-Integrative, axé sur les protéines intrinsèquement désordonnées (IDP), afin d'examiner en détail les capacités de chaque modèle à modéliser les conformations désordonnées. Les résultats ont mis en évidence des différences marquées entre les modèles.
Comme le montre la figure ci-dessous, sur l'ensemble de données PeptoneDB-CS, les performances de chaque modèle varient considérablement en fonction du degré de désordre des protéines (score G) : ESMFold et ESMFlow sont précis dans la prédiction des régions ordonnées, mais leurs performances sont considérablement réduites dans les régions désordonnées ; IDP-o présente un schéma complémentaire typique : plus le degré de désordre est élevé, meilleures sont les performances.PepTron maintient une stabilité élevée sur l'ensemble du spectre conformationnel ordonné-désordonné.Cette capacité d'équilibrage a été validée plus en détail dans l'ensemble de données PeptoneDB-SAXS et l'analyse pondérée ultérieure, démontrant que PepTron peut capturer efficacement la diversité conformationnelle des protéines désordonnées sans sacrifier la précision des structures ordonnées.
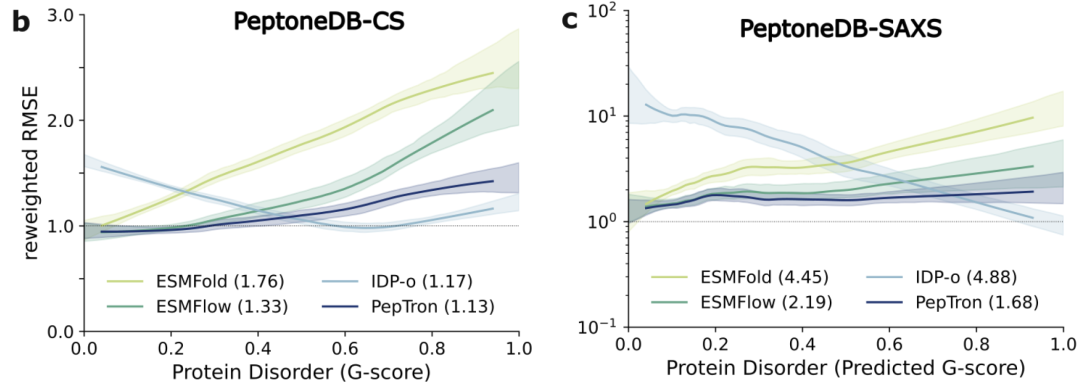
D'autres résultats de comparaison inter-modèles sont présentés dans la figure ci-dessous. Bien qu'AlphaFold2 et Boltz2 restent les modèles dominants pour la prédiction des protéines ordonnées, leurs performances diminuent systématiquement à mesure que le degré de désordre augmente ; en revanche,PepTron et BioEmu présentent une plus grande robustesse sur l'ensemble du spectre conformationnel, ce qui les rend plus adaptés à la gestion des caractéristiques structurelles très hétérogènes des IDP.
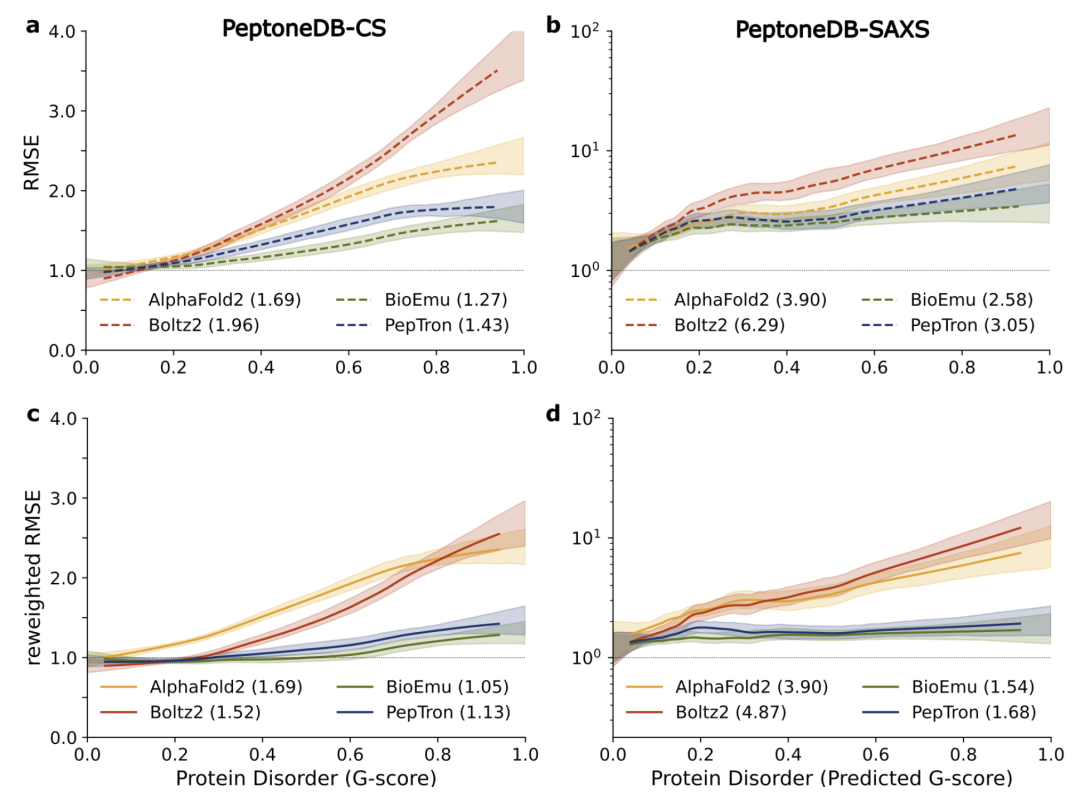
Afin de s'assurer que l'entraînement sur des régions désordonnées n'altérait pas sa capacité prédictive pour les protéines ordonnées, l'équipe de recherche a également mené des tests supplémentaires sur des données de structure ordonnée issues de CAMEO22 et CASP14. Les résultats ont montré que…PepTron affiche des performances conformes à celles d'ESMFlow sur des indicateurs clés tels que RMSD, LDDT et TM, démontrant qu'il ne compromet pas la précision des structures ordonnées tout en étendant les capacités de modélisation IDR.
Dans l'ensemble de données PeptoneDB-Integrative, qui intègre plusieurs métriques expérimentales, comme illustré dans la figure ci-dessous, les performances du modèle révèlent des différences supplémentaires. IDP-o se distingue particulièrement après une pondération par entropie maximale, surpassant significativement les autres modèles en termes d'erreur quadratique moyenne (RMSE) et de facteur Q du RDC. PepTron et BioEmu présentent des valeurs similaires pour les métriques RDC, mais BioEmu est plus performant pour la prédiction des déplacements chimiques locaux. Il est à noter que même sans pondération,IDP-o continue de dominer la plupart des indicateurs locaux et globaux, démontrant ainsi son avantage naturel dans la couverture des conformations protéiques désordonnées.
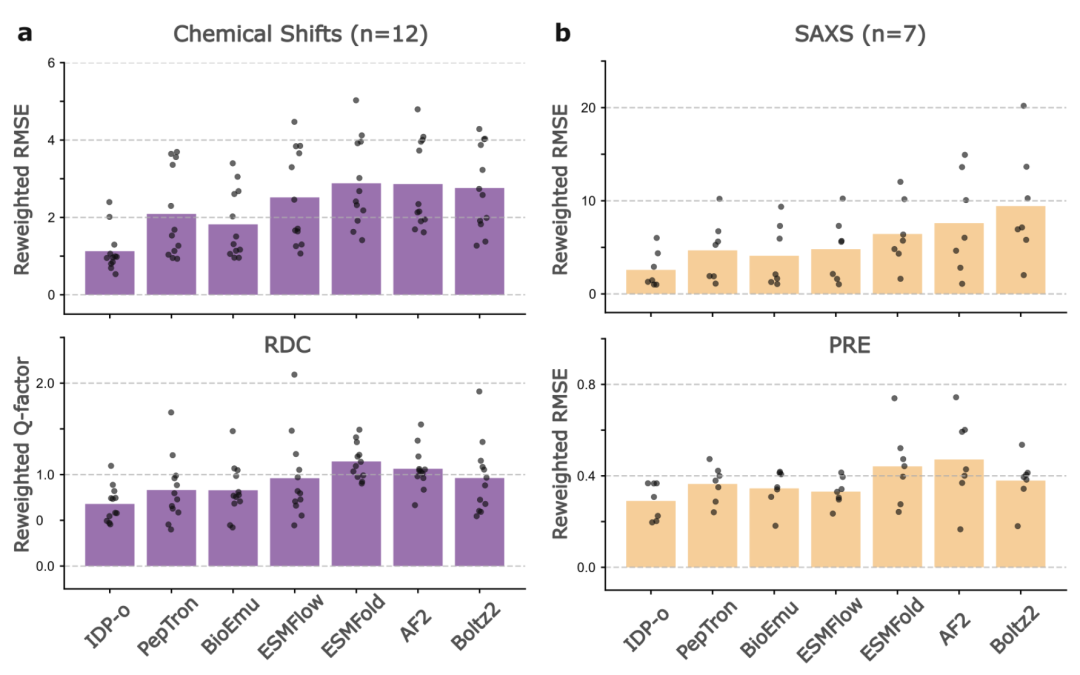
L'étude a également mis en évidence plusieurs points faibles communs au modèle actuel :La plupart des modèles ne parviennent pas à saisir les préférences de contact à longue portée et présentent divers degrés de biais de structure secondaire. De plus, les modèles classiques ont généralement du mal à décrire avec précision l'état déplié des « séquences repliées conditionnellement », tandis que l'IDP-o démontre une supériorité unique à cet égard.
Du désordre à l'ordre : avancées mondiales et nouveaux chapitres de la recherche sur les personnes déplacées
Les protéines intrinsèquement désordonnées (IDP) deviennent rapidement un domaine de recherche de pointe dans les sciences de la vie et les industries pharmaceutiques mondiales en raison de leurs caractéristiques conformationnelles très dynamiques et de leur association étroite avec de nombreuses maladies majeures.
Dans le milieu universitaire, la technologie de prédiction de structure par IA devient un outil essentiel pour décrypter le « mot de passe dynamique » des fournisseurs d'identité.La méthode AlphaFold-Metainference proposée par l'Université de Cambridge,En combinant les cartes d'erreur d'alignement AlphaFold avec des simulations de dynamique moléculaire, cette approche surmonte la limitation de l'AlphaFold traditionnel, qui prédit principalement des structures stables, et construit avec succès des IDP et des ensembles de structures contenant des régions désordonnées, offrant une nouvelle voie pour comprendre leur polymorphisme.
Titre de l'article :
Prédiction par AlphaFold d'ensembles structuraux de protéines désordonnées
Lien vers l'article :https://www.nature.com/articles/s41467-025-56572-9
L'équipe de l'Université de Copenhague a par ailleurs intégré AlphaFold à un modèle de langage protéique.Elle a permis de prédire à grande échelle la conformation du protéome désordonné humain.Cela démontre l'universalité et l'évolutivité de la technologie d'IA dans la recherche sur les personnes ayant une déficience intellectuelle.
Titre de l'article :
Ensembles conformationnels du protéome intrinsèquement désordonné humain
Lien vers l'article :https://www.nature.com/articles/s41586-023-07004-5
La capacité des découvertes universitaires à véritablement transformer le traitement des maladies dépend de la capacité de l'industrie à traduire les technologies en applications concrètes. La collaboration entre la société de biotechnologie britannique Peptone et la société pharmaceutique allemande Evotec…Cela démontre qu'il existe une voie possible pour étendre la recherche sur les IDP au développement de médicaments.Grâce à la plateforme de spectrométrie de masse par échange hydrogène-deutérium (HDX-MS) ultrarapide de Peptone, les chercheurs peuvent suivre en temps réel l'évolution dynamique des protéines désordonnées et identifier des sites de liaison difficiles à localiser par les méthodes de détermination structurale classiques. Combinée aux atouts d'Evotec en matière de validation de cibles, de criblage de médicaments et de développement clinique, cette approche permet de transformer des cibles IDP difficiles à cibler en molécules candidates à potentiel thérapeutique.
Cette série d'avancées confirme non seulement la tendance du modèle PepTron qui « couvre tout le spectre des structures ordonnées et désordonnées », mais elle témoigne également du fait que les protéines désordonnées, autrefois considérées comme difficiles à appréhender, deviennent progressivement des cibles clés en médecine de précision et en biopharmacie. Grâce aux percées technologiques continues et à l'approfondissement de la collaboration industrielle, les protéines intrinsèquement désordonnées (IDP) pourraient offrir un cadre entièrement nouveau pour la compréhension et les voies d'intervention dans le traitement des maladies futures.
Liens de référence :
1.https://www.vbdata.cn/intelDetail/717834
2.https://c.m.163.com/news/a/JDIR2LQJ0552ZPM2.html
3.https://www.vbdata.cn/intelDetail/580634








