Command Palette
Search for a command to run...
Soutenant La génération/le repliement/le Repliement Inverse Des Protéines, l'Université Du Hunan/Université De l'Académie Chinoise Des sciences/ByteDance a Proposé Le Modèle APM Pour Réaliser Une Conception tout-atomique Et Une Optimisation Fonctionnelle
Principales exécutantes du vivant, les protéines accomplissent souvent leurs fonctions sous forme de complexes multichaînes. De la reconnaissance anticorps-antigène à la liaison enzyme-substrat, l'interaction précise entre protéines multichaînes est au cœur de la compréhension du mécanisme du vivant.Cependant, la modélisation actuelle des protéines basée sur l'IA présente un biais significatif en faveur des protéines monocaténaires. Bien que des modèles tels qu'AlphaFold et la série ESM aient permis des avancées majeures dans le repliement et la conception de protéines monocaténaires, la modélisation des complexes multicaténaires en est encore à ses balbutiements.
Les méthodes existantes de traitement des protéines multi-chaînes adoptent généralement la stratégie de « connexion de pseudo-séquence », obligeant les multi-chaînes à être traitées comme des chaînes simples.Cette méthode limite considérablement l'expression naturelle des interactions interchaînes. Dans les complexes biologiques réels, les interactions au niveau atomique entre les positions spatiales des interchaînes et l'interface de liaison (telles que les liaisons hydrogène et les interactions hydrophobes) ne peuvent être modélisées avec précision par des connexions linéaires. De plus, la génération de structures entièrement atomiques se heurte à un double défi : la conformation complexe des chaînes latérales des acides aminés et la forte dépendance séquence-structure font de la conception de novo de complexes multichaînes un problème complexe sur le terrain.
Pour combler cette lacune de recherche, l'Université du Hunan, l'Université de l'Académie chinoise des sciences et l'équipe ByteDance Seed ont proposé APM (All-Atom Protein Generative Model), un modèle de génération de protéines tout atome conçu spécifiquement pour les complexes protéiques multi-chaînes. L'APM peut non seulement générer directement des complexes multi-chaînes avec des structures entièrement atomiques, mais prend également en charge des tâches de base telles que le repliement et le repliement inverse, et démontre d'excellentes performances dans la conception de protéines fonctionnelles telles que les anticorps et les peptides.
Les résultats de la recherche ont été sélectionnés pour l'ICML 2025 sous le titre « Un modèle génératif tout-atome pour la conception de complexes protéiques ».
Points saillants de la recherche :
* Modélisation native multi-chaînes : abandonnez les connexions pseudo-séquences et apprenez directement les interactions au niveau atomique entre la distribution spatiale indépendante des multi-chaînes et l'interface de liaison ;
* Optimisation de la représentation de tous les atomes : équilibrer l'efficacité du calcul et les détails structurels, et réaliser la génération de structures au niveau atomique grâce à la représentation conjointe du type d'acide aminé, de la structure principale et de l'angle de torsion de la chaîne latérale ;
* Renforcement de la dépendance séquence-structure : Maintenir l'association profonde entre séquence et structure en découplant le processus de bruit et l'entraînement aux tâches bidirectionnelles (pliage/dépliage).

Adresse du document :
Suivez le compte officiel et répondez « APM » pour obtenir le PDF completEnsemble de données de génération de protéines APM :
Autres articles sur les frontières de l'IA :
Ensemble de données : échantillons riches allant d'une chaîne unique à plusieurs chaînes
APM est formé sur la base d'un ensemble de données protéiques multi-sources soigneusement construit qui intègre les informations de structure et de séquence des protéines à chaîne unique et à chaînes multiples, fournissant ainsi de riches supports d'apprentissage pour le modèle.
L'ensemble de données mono-chaîne constitue une base solide pour la modélisation en chaîne grâce à la fusion multi-sources et au filtrage de qualité. Il contient 187 494 échantillons au total, couvrant un large éventail de types de protéines et de catégories fonctionnelles. Ses données proviennent principalement de trois bases de données faisant autorité :
* Base de données PDB : Après le processus de traitement des données MultiFlow, 18 684 échantillons ont été examinés ;
* Base de données Swiss-Prot : structures sélectionnées de haute qualité avec pLDDT>85 et 140 769 échantillons obtenus ;
* Base de données AFDB : En utilisant des critères de sélection plus stricts, les échantillons avec pLDDT> 95 ont été conservés, totalisant 28 041 échantillons.
L'ensemble de données sur les protéines multichaînes contient un total de 11 620 échantillons, couvrant des complexes protéiques de 2 à 6 chaînes, fournissant des données essentielles à la modélisation multichaîne. Ces données proviennent des données d'assemblage biologique de la base de données PDB (Biological Assemblies). Afin d'éviter toute fuite d'informations lors des tâches en aval, l'équipe de recherche a exclu trois types d'échantillons : les échantillons présents dans la base de données d'anticorps SAbDab ; les échantillons contenant des chaînes d'une longueur inférieure à 30 (considérées comme des peptides) ; et les échantillons d'une longueur supérieure à 2 048 ou sans identifiant de cluster.
Pour améliorer la capacité de généralisation du modèle, les chercheurs ont découpé de manière aléatoire les échantillons multi-chaînes au cours du processus de formation : pour les échantillons contenant plus de 384 résidus, les 384 acides aminés les plus proches ont été conservés, centrés sur les paires de résidus à l'interface de liaison inter-chaîne.Cette stratégie d’élagage garantit que le modèle peut se concentrer sur les zones de liaison clés tout en évitant les problèmes de dépassement de mémoire.De plus, les chercheurs ont combiné proportionnellement des données mono-chaînes et multi-chaînes, exploitant la richesse des données mono-chaînes pour améliorer les capacités de modélisation intra-chaîne. Chaque point d'échantillonnage est associé à des métadonnées riches, notamment la localisation géographique (site d'interaction inter-chaînes), les propriétés structurelles (comme le type de structure secondaire) et les caractéristiques de la séquence (type et conservation des acides aminés). Ces informations fournissent des indices multidimensionnels permettant au modèle d'identifier les relations entre séquence, structure et fonction.
Ensemble de données de génération de protéines APM :
Architecture du modèle : un cadre collaboratif de génération d'atomes entiers en trois modules
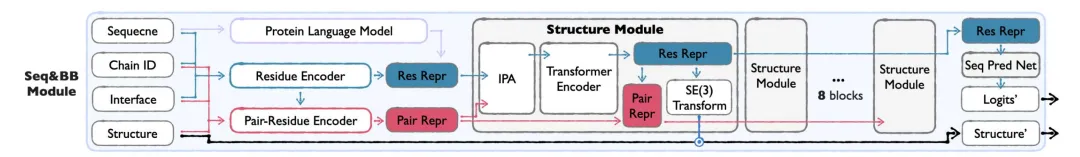
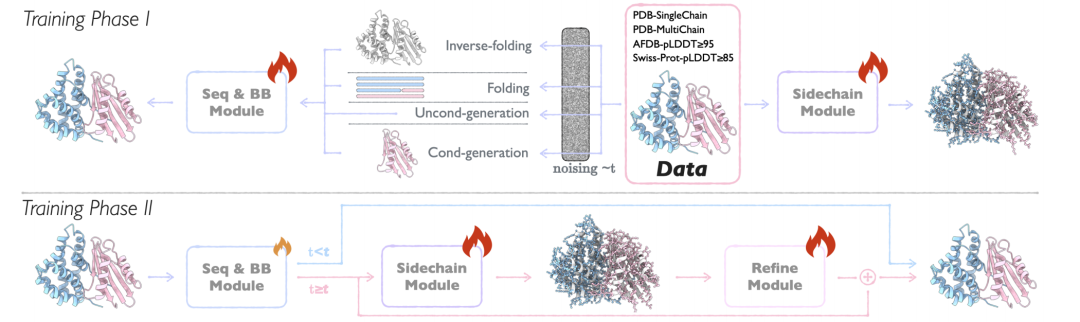
L'architecture principale d'APM est composée de trois modules aux fonctions claires : le module de génération de séquence et de backbone (module Seq&BB), le module de génération de sidechain (module Sidechain) et le module d'affinage.Grâce à une conception innovante, la génération de bout en bout, de la séquence à la structure entièrement atomique, est réalisée, tout en prenant en charge diverses tâches de conception de protéines multi-chaînes.
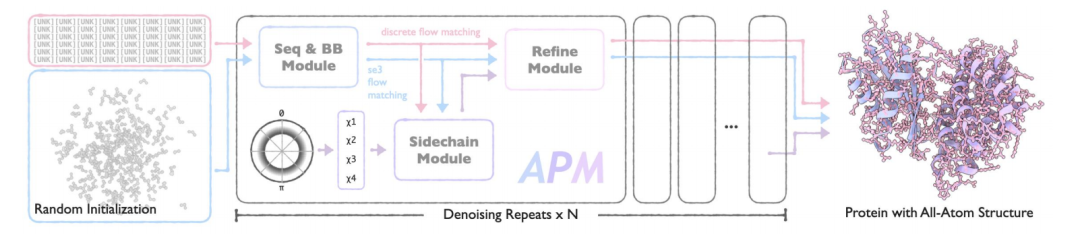
Module Seq&BB
Ce module constitue la base de l'APM. Il adopte la méthode de correspondance de flux pour réaliser la génération conjointe de séquences et de squelettes protéiques, et peut gérer des tâches de modélisation collaborative séquence-structure au niveau des résidus.En découplant le processus de bruit de séquence et de structure, l'altération de la relation de dépendance séquence-structure est réduite, et les tâches de repliement/repliement inverse sont réalisées avec une probabilité de 50% afin de renforcer l'apprentissage bidirectionnel des dépendances. L'innovation principale du module est :
* Processus de découplage du bruit :La séparation des processus de bruit de séquence et de structure évite la destruction des dépendances intermodales dans les méthodes traditionnelles. La séquence de bruit et le noyau de bruit sont échantillonnés indépendamment à différents pas de temps, ce qui permet au modèle d'apprendre les dépendances bidirectionnelles séquence-structure.
* Correspondance de flux SE(3) :Compte tenu des caractéristiques de transformation spatiale de la chaîne principale des protéines, une correspondance de flux de groupe euclidien spécial tridimensionnel (SE(3)) est introduite pour gérer séparément les parties de translation et de rotation.
* Apprentissage multitâche :Il prend également en charge les tâches de génération inconditionnelle, de génération conditionnelle, de repliement et de repliement inverse, et améliore la capacité de généralisation du modèle grâce à l'apprentissage de tâches mixtes. La fonction de perte inclut la perte de correspondance de flux et la perte de cohérence pour garantir la fluidité de la trajectoire générée.
Module Sidechain
Pour obtenir une génération de structure entièrement atomique, le module Sidechain prédit la conformation des chaînes latérales d'acides aminés en fonction de la séquence et du squelette générés par Seq&BB.
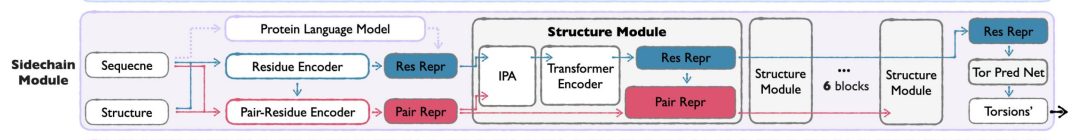
Le module adopte les stratégies suivantes :
* angle de torsion signifie :La structure de la chaîne latérale est paramétrée par les angles de torsion de la chaîne latérale (jusqu'à 4 liaisons rotatives), équilibrant l'efficacité de calcul et les détails au niveau atomique, évitant la complexité de la modélisation directe des coordonnées de tous les atomes.
* Formation en deux étapes :La première étape se concentre sur la tâche d'emballage de la chaîne latérale et apprend la distribution des conformations réelles de la chaîne latérale ; la deuxième étape passe à la reconstruction des chaînes latérales réelles à partir des structures prédites pour garantir l'applicabilité du modèle dans le scénario de génération.
* Conception légère :Comparé au module Seq&BB, le module Sidechain utilise moins de blocs structurels et des dimensions cachées plus petites.
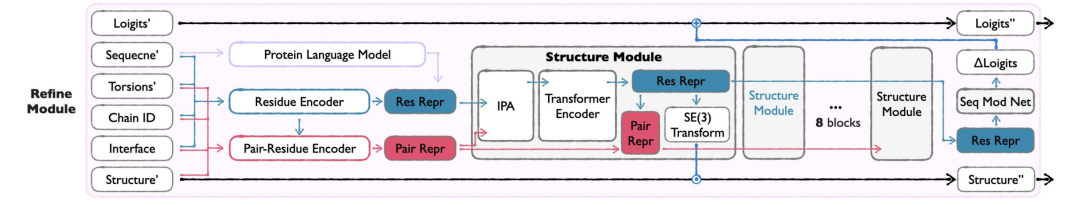
Module d'affinage
En tant que dernier maillon de l'APM, le module Refine intègre la sortie du module Seq&BB et Sidechain, optimise la séquence et la dorsale en corrigeant la perte, réduit les conflits atomiques et améliore la rationalité structurelle.Les informations atomiques complètes sont utilisées pour optimiser la séquence et la structure de la chaîne principale, résoudre les conflits structuraux et rapprocher les résultats générés de la protéine naturelle. Ce module n'est activé qu'en fin de génération (t≥0,8) afin de garantir une qualité d'entrée suffisante pour soutenir l'optimisation.
Conclusion expérimentale : Vérification multidimensionnelle des performances révolutionnaires de l'APM
La vérification expérimentale d'APM couvre les tâches de base à chaîne unique, les tâches de base à chaînes multiples et la conception fonctionnelle en aval, et les résultats sont tous excellents.
Tâche sur les protéines à chaîne unique : capacités de base comparables aux modèles professionnels
Dans la tâche de pliage, sur l'ensemble de données PDB,Le RMSD de l'APM est de 4,83/2,64,Le score TM a atteint 0,86/0,91, ce qui est comparable aux performances d'ESM3, MultiFlow et d'autres modèles ; dans la tâche de pliage inverse, le taux de récupération des acides aminés (AAR) a atteint 50,44%, dépassant les 46,58% de ProteinMPNN.
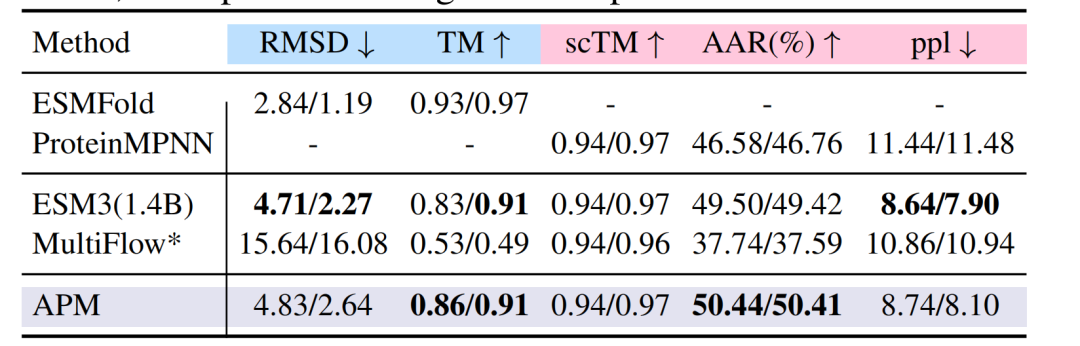
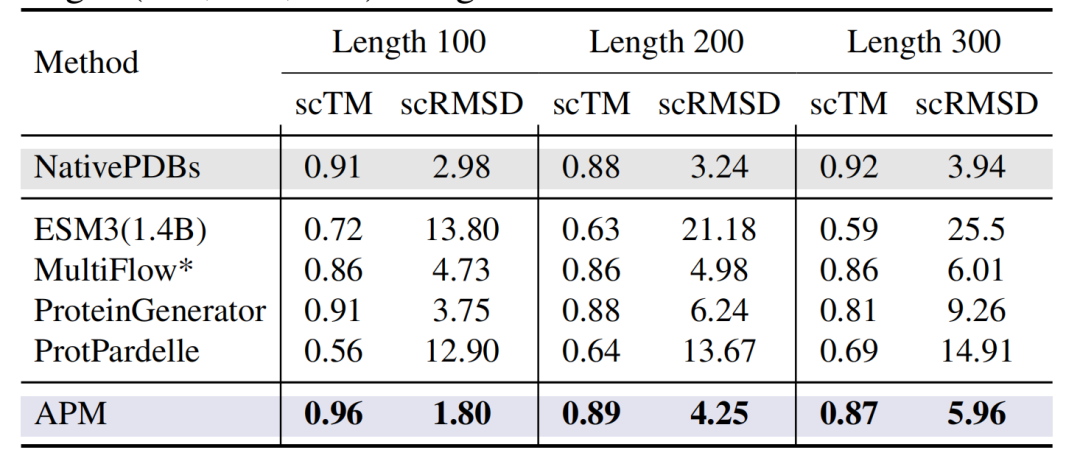
De plus, comme le montre la figure ci-dessous, dans les protéines générées inconditionnellement avec des longueurs de 100 à 300 résidus,Le scTM de l'APM est aussi élevé que 0,96 (longueur 100) et le scRMSD est aussi bas que 1,80.Significativement meilleur que les modèles de conception entièrement atomique tels que ESM3 (1.4B) et ProtPardelle.
Tâches protéiques multi-chaînes : l'avantage principal de la modélisation native
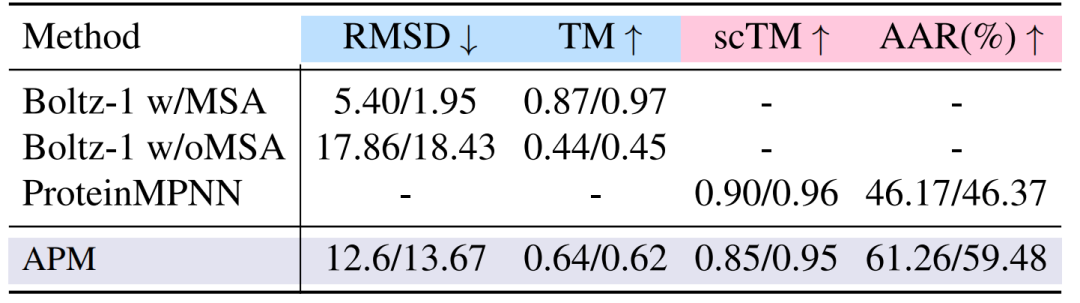
Dans les expériences de pliage et de dépliage,Sur le complexe 2-6 chaînes, la performance de repliement de l'APM est de 12,6/13,67, ce qui est inférieur à celui de Boltz-1, mais nettement supérieur à celui de Boltz-1 sans MSA ; le repliement inverse du scTM atteint 0,85/0,95, proche de celui de Boltz-1 avec MSA, prouvant la validité de l'association séquence-structure. Les résultats expérimentaux sont présentés dans la figure ci-dessous.
Deuxièmement,Le complexe multi-chaînes a une forte affinité de liaison.En prenant comme exemple une longueur de chaîne de 50 à 100, l'énergie de liaison ΔG_RAA après relaxation de tous les atomes atteint -112,65/-116,98, ce qui est nettement meilleur que Chroma (-83,96/-86,66) et APM_BB (-114,94/-114,45) en utilisant uniquement la chaîne principale, prouvant la nécessité d'informations sur tous les atomes pour modéliser les interactions inter-chaînes.
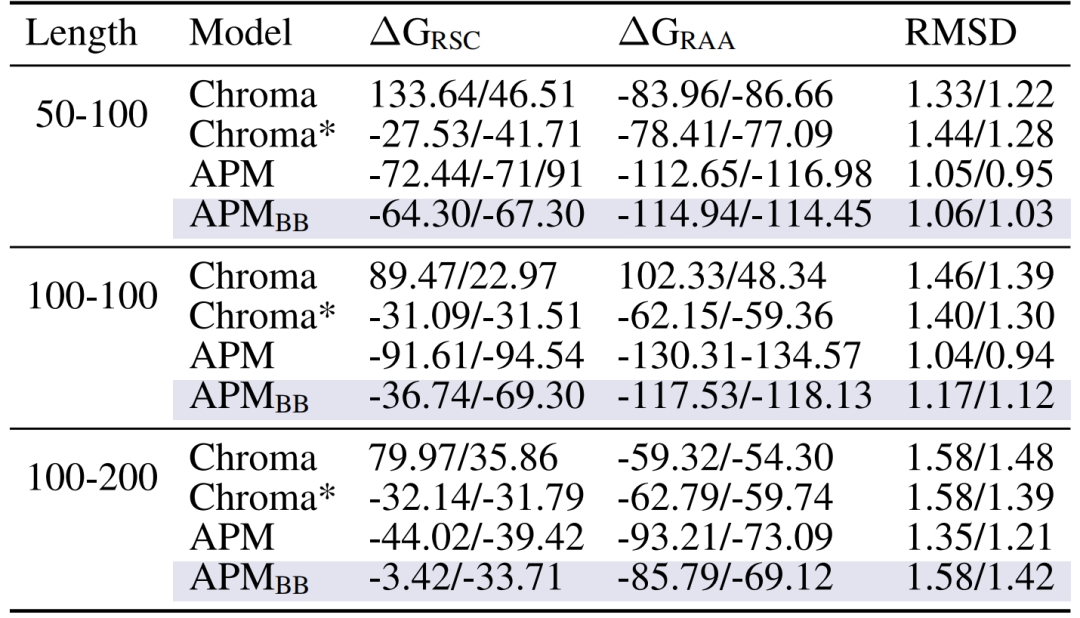
Affinité de liaison interchaîne entre les complexes générés
Conception fonctionnelle en aval : avancées dans l'application des anticorps et des peptides
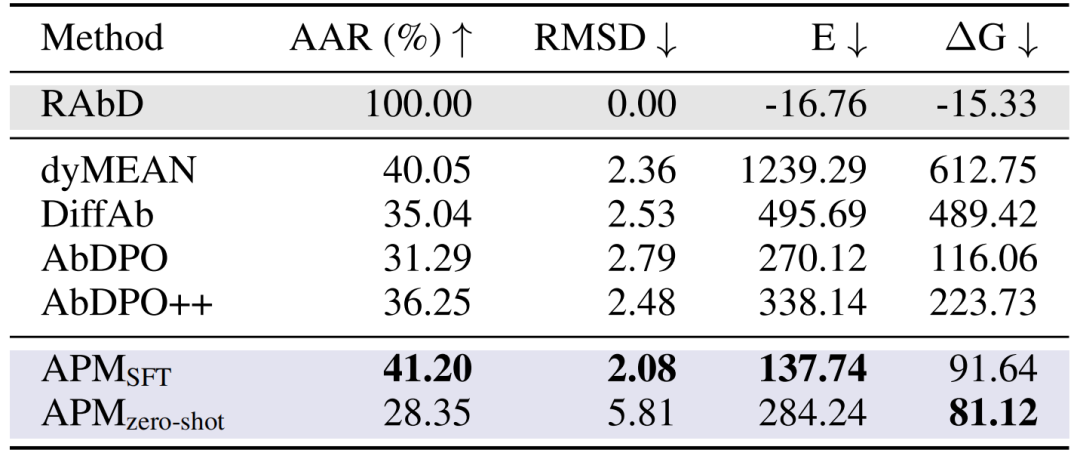
Conception de l'anticorps CDR-H3 :Lors du test de référence RAbD, l'AAR de l'APM a atteint 41,20%, l'écart type RMSD était de 2,08 et l'énergie de liaison ΔG était de 91,64, surpassant des méthodes telles que dyMEAN et DiffAb. Bien que la séquence de l'anticorps générée par l'échantillon zéro soit très différente de la séquence naturelle, l'énergie de liaison est meilleure (ΔG 81,12), prouvant sa capacité de liaison universelle.
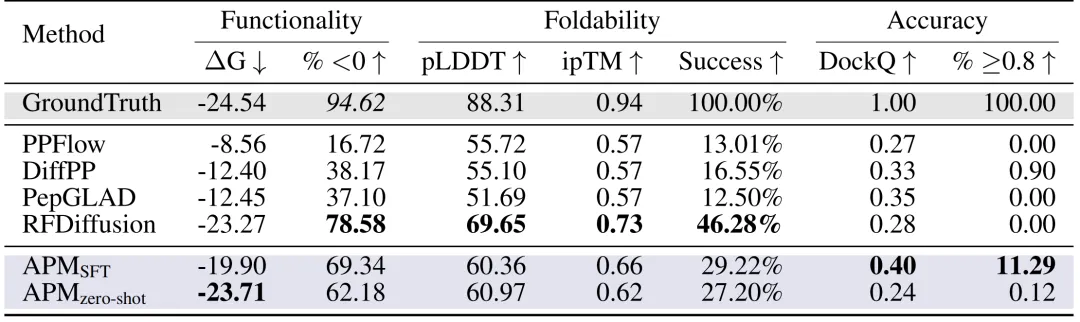
Conception de peptides :Sur les ensembles de données PepBench et LNR, les chercheurs ont évalué de manière exhaustive les méthodes de conception de peptides sous trois aspects clés : fonctionnalité, aptitude au pliage et précision. Comme le montre la figure ci-dessous, l'énergie de liaison ΔG de l'APM (SFT) a atteint -19,90, 69,341 échantillons TP3T avaient ΔG < 0, et la proportion de DockQ ≥ 0,8 a atteint 11,291 TP3T, dépassant largement PPFlow, PepGLAD et d'autres méthodes. La stabilité du pliage (pLDDT 60,36, ipTM 0,66) était excellente.
La collaboration entre l'industrie et la R&D permet des avancées dans la technologie de génération de protéines entièrement atomiques
Dans le domaine biologique de pointe de la génération de protéines entièrement atomiques, les milieux universitaires et commerciaux n'ont jamais cessé de l'explorer, et une série de résultats révolutionnaires continuent d'attirer l'attention.
Dans le monde académique, AlphaFold3 lancé par l'équipe DeepMind a démontré de fortes capacités dans le domaine de la génération de protéines tout-atome en intégrant des informations structurelles multi-échelles avec des données de séquence évolutive.A obtenu une modélisation précise des modèles complexes de repliement des protéines,Notamment pour la génération de complexes tout-atomiques contenant des cofacteurs et des ions métalliques, la précision structurale et la rationalité énergétique ont été considérablement améliorées par rapport aux méthodes traditionnelles. Développé par l'équipe de recherche de l'Université de Stanford, ESM-IF1 adopte une approche différente. S'appuyant sur un modèle de repliement implicite entraîné à partir de données massives de séquences évolutives, il peut générer directement des structures protéiques tout-atomiques présentant des caractéristiques conformationnelles naturelles et se montre remarquablement performant dans la construction précise de centres actifs enzymatiques.
Le monde des affaires se déploie également activement dans ce domaine, favorisant les applications industrielles grâce à l'innovation technologique. Beijing Bio-Geometry Biotechnology Co., Ltd. a lancé le premier modèle protéique atomique complet au monde : GeoFlow V2. Ce modèle a construit un cadre de génération de diffusion de bout en bout permettant une régulation précise des atomes de protéines. Dans la conception atomique complète des régions CDR des anticorps,Il peut optimiser l’affinité et la stabilité en même temps, améliorant considérablement l’efficacité du développement de médicaments.Insilico Medicine, société de biotechnologie américaine, a développé un système de génération de protéines axé sur la conception de protéines cibles pour médicaments. La stratégie de génération multi-contraintes adoptée permet d'optimiser les sites de liaison entre les protéines et les petites molécules médicamenteuses, tout en garantissant la rationalité de la structure tout-atomique, offrant ainsi une base solide pour un criblage efficace des médicaments candidats.
Ces avancées théoriques dans le monde universitaire et ces innovations d’application dans le monde des affaires,Ensemble, nous allons faire passer la technologie de génération de protéines entièrement atomiques du laboratoire à la pratique industrielle, en fournissant un soutien essentiel aux avancées dans le développement de médicaments de précision, la conception de nouveaux biocatalyseurs et la biologie synthétique, et nous espérons créer une valeur énorme dans le traitement des maladies et la biofabrication à l'avenir.
Liens de référence :
1.https://mp.weixin.qq.com/s/a0bl9ek90t_-y8wy69Yu6Q
2.https://mp.weixin.qq.com/s/P-5o-R1qZY52Pq1yK5j6cQ