Command Palette
Search for a command to run...
L'équipe Du MIT Ouvre Les Sources De BoltzGen, Permettant La Conception De Liants Protéiques Sur Tous Les Types Moléculaires, Atteignant Une Affinité Nanomolaire Pour La Cible 66%.

Dans les domaines de la découverte de médicaments et de l'ingénierie biomoléculaire, la conception de novo de liants est une méthode essentielle pour la découverte automatisée de médicaments. Grâce à la simulation numérique et à l'apprentissage profond, les chercheurs peuvent générer des structures peptidiques ou protéiques capables de se lier à des cibles spécifiques, permettant ainsi le développement de nouvelles modalités médicamenteuses telles que les anticorps, les nanocorps et les peptides cycliques.
Cependant, les stratégies traditionnelles de conception de protéines reposent principalement sur des calculs physiques tels que des simulations de dynamique moléculaire et des algorithmes d'optimisation de séquences. Bien qu'une haute précision puisse être obtenue dans un seul système,Cependant, le coût de calcul est élevé, l’espace de conception est limité et il est difficile de traiter simultanément des cibles multimodales telles que des protéines, des petites molécules et de l’ARN.Bien que les modèles génératifs profonds actuels aient amélioré leur vitesse de génération dans une certaine mesure, ils manquent généralement de capacités de raisonnement structurel à l'échelle atomique et sont optimisés pour des catégories spécifiques de molécules, ce qui limite leur polyvalence. De plus, l'évaluation des modèles repose souvent sur des complexes similaires existants dans l'ensemble d'apprentissage, ce qui complique la vérification des capacités de généralisation pour des cibles « invisibles ». Ils manquent de mécanismes de génération contrôlables et d'expressions de contraintes structurelles flexibles, ce qui limite l'efficacité et l'interprétabilité de la conception.
Pour résoudre ce problème,Le MIT, en collaboration avec Boltz et d'autres institutions, a proposé le « modèle génératif tout-atome » BoltzGen, qui unifie la prédiction de structure et la conception complexe.Ce modèle remplace non seulement les étiquettes de résidus discrètes traditionnelles par des représentations géométriques continues pour obtenir une formation conjointe du repliement des protéines et de la conception de liaison dans un seul système, mais construit également un « langage de spécification de conception » flexible pour obtenir une génération contrôlable sur tous les types moléculaires.
Les résultats expérimentaux montrent queLes conceptions de nanocorps et de conjugués protéiques de BoltzGen ont toutes pour objectif d'atteindre une affinité nanomolaire pour 66%.Pour la première fois, il a été démontré qu'un « système modèle unique » peut atteindre une optimisation simultanée des performances de repliement et de liaison dans la conception de biomolécules multimodales.
Actuellement, les résultats de recherche pertinents ont été publiés sous le titre « BoltzGen : Toward Universal Binder Design ».
Adresse GitHub :
https://github.com/HannesStark/boltzgen
Points saillants de la recherche :
* Prédiction de structure unifiée et conception de liant dans un seul modèle génératif entièrement atomique, permettant le repliement simultané des protéines, la modélisation du site de liaison et la génération de séquences avec une précision au niveau atomique, améliorant considérablement la rationalité physique et la contrôlabilité de la conception moléculaire ;
* Un « langage de spécification de conception » universel est proposé, permettant au modèle de basculer de manière flexible entre différents systèmes tels que les protéines, les nanoanticorps, les peptides cycliques et les petites molécules, réalisant une génération de structure intermodale et un contrôle des contraintes, et élargissant le champ d'application de l'IA générative dans le domaine de la conception biomoléculaire.

Adresse du document :
https://go.hyper.ai/3sx2K
Suivez le compte officiel et répondez « BoltzGen » pour obtenir le PDF complet
Autres articles sur les frontières de l'IA :
Ensembles de données mixtes : stratégies de formation multimodales
L'équipe de recherche a adopté un cadre de formation conjointe multi-niveaux et intermodal lors de la formation de BoltzGen.Les principales sources des ensembles de données utilisés comprennent trois catégories :
* Structures expérimentales de haute qualité provenant de la Protein Data Bank (PDB), couvrant une variété de structures complexes telles que l'ARN, l'ADN et les petites molécules de protéines, fournissant des contraintes de liaison chimique réalistes et des données de distribution géométrique tridimensionnelle pour le modèle ;
* Données expérimentales de la base de données AlphaFold (AFDB), prédites et réapprises par AlphaFold2, couvrant des modèles de pliage fiables générés par des expériences ;
* Les échantillons de structure composite générés par le modèle Boltz-1 couvrent des scénarios multimodaux tels que la liaison de petites molécules et les interactions ARN-ADN, ce qui peut améliorer la capacité de généralisation du modèle sur différents types de biomolécules.
Afin d'éviter que le modèle ne soit trop biaisé en faveur de types structuraux spécifiques, l'équipe de recherche a éliminé les ensembles de données suréchantillonnés pour les anticorps et les TCR afin de préserver la diversité de l'espace généré. De plus, tous les échantillons structuraux ont été recadrés aléatoirement et traités de manière multitâche pendant l'entraînement, permettant au modèle de gérer aléatoirement des tâches telles que la prédiction du repliement, la conception complexe et la finalisation de la structure à chaque itération d'entraînement. Ce cadre d'apprentissage unifié et multifonctionnel permet au modèle de générer des structures à l'échelle atomique tout en offrant des capacités de compréhension intermodale.
Architecture du modèle : inférence sur tous les atomes, du bruit à la structure
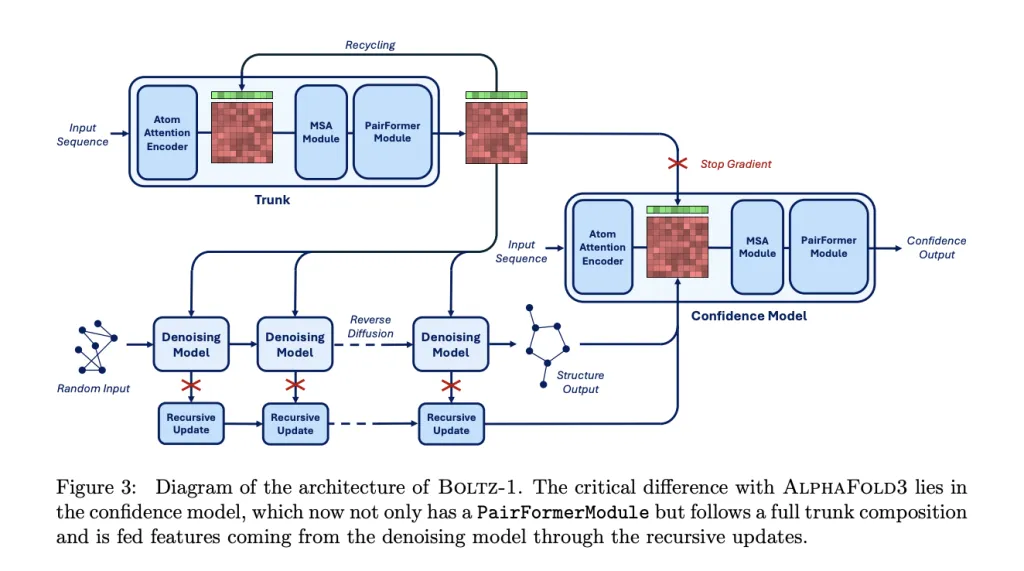
Le modèle conserve les principaux composants des architectures AlphaFold3 et Boltz-2 et apporte quelques améliorations sur cette base pour introduire davantage d'entrées conditionnelles.
Comme le montre la figure ci-dessous, l’ensemble du modèle est divisé en deux parties principales :Un Trunk plus grand (réseau fédérateur) et un module de diffusion (module de diffusion).Le module Trunk génère des représentations de jetons et de paires pour le contrôle conditionnel, tandis que le module Diffusion génère la structure 3D à partir de ces représentations. Le module Trunk ne s'exécute qu'une seule fois, tandis que le module Diffusion effectue plusieurs itérations pour débruiter progressivement les coordonnées 3D de tous les atomes.
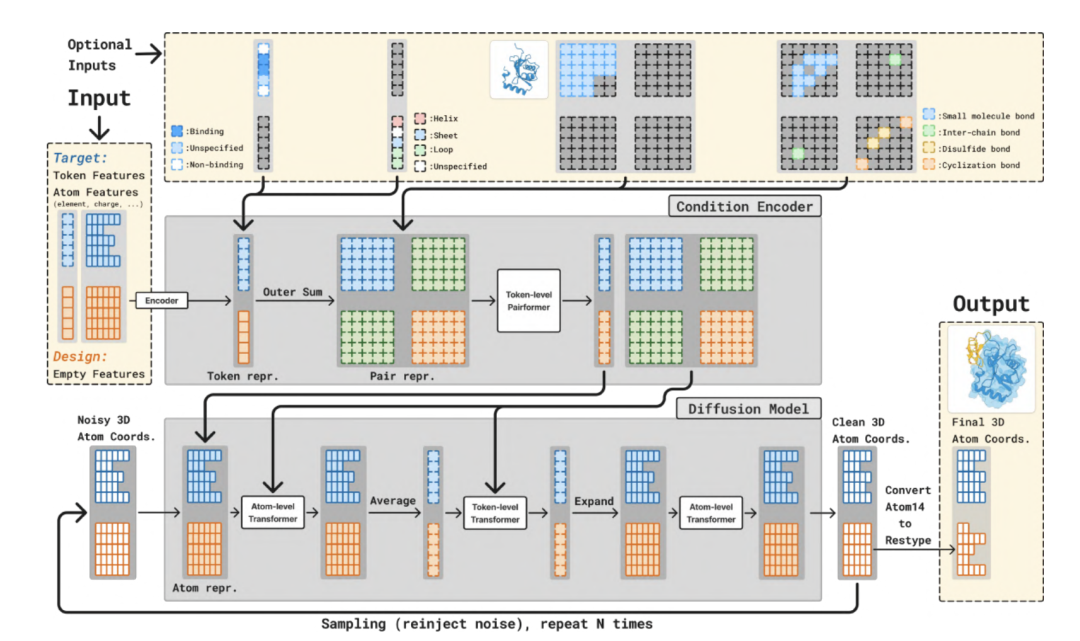
Au stade Trunk, il est similaire au module Trunk de Boltz-2 et est responsable de l'analyse de la structure de la protéine d'entrée et des informations cibles. Le module Trunk traite les structures moléculaires tokenisées.Le cadre principal utilise une architecture PairFormer, utilisant Triangle Attention pour modéliser efficacement les relations spatiales entre les atomes. Associé au codage géométrique des résidus, il déduit simultanément les types de résidus et les coordonnées atomiques dans un espace continu, éliminant ainsi le recours à des étiquettes d'acides aminés discrètes. Ce mécanisme permet au modèle de comprendre véritablement les lois physiques de la structure dès la génération, plutôt que de se fier uniquement à la mémorisation des données.
Au stade du module de diffusion,Ce module reçoit des coordonnées atomiques 3D bruyantes en entrée.et prédit ses coordonnées débruitées. Il utilise une architecture Transformer standard, fonctionnant à la fois au niveau atomique et au niveau du jeton. BoltzGen utilise un modèle de diffusion en espace continu pour débruiter progressivement les coordonnées atomiques. Il prédit les vecteurs de bruit pour transformer les états initiaux aléatoires en conformations stables, préservant ainsi les contraintes de la surface d'énergie moléculaire pendant le processus de génération afin d'éviter les conflits physiques ou l'effondrement structurel.
Résultats expérimentaux : Validation de la conception universelle sur 26 cibles
Dans la partie expérimentale, la vérification des performances du modèle BoltzGen a couvert plusieurs dimensions allant des protéines aux peptides, des nouveaux agents pathogènes aux cibles de petites molécules, démontrant une excellente généralisation et contrôlabilité.
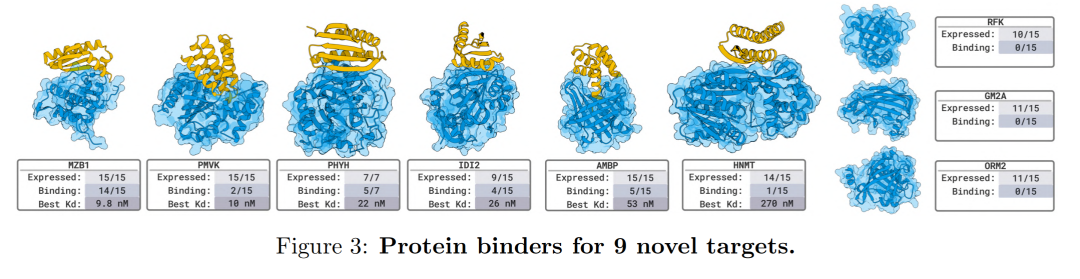
L’équipe a testé un total de 26 cibles dans 8 projets indépendants de validation en laboratoire humide.Les résultats ont porté sur divers types de liaison, notamment des nanocorps, des protéines et des peptides linéaires et cycliques. BoltzGen a maintenu un taux de réussite élevé contre des cibles complexes et inédites : lors de neuf expériences avec de nouvelles cibles totalement différentes des données d'entraînement, les protéines et les nanocorps conçus ont tous atteint une liaison nanomolaire (nM) de haute affinité à la cible 66%, démontrant ainsi la puissance du raisonnement structurel et les capacités de conception intermodale du modèle.
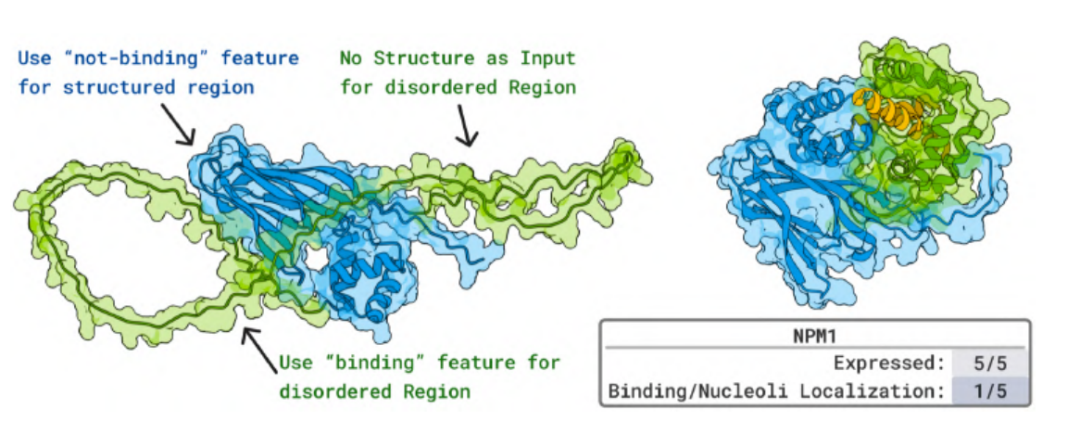
Dans des expériences sur des peptides bioactifs aux structures diverses,Les protéines conçues par BoltzGen peuvent se lier à différents types de molécules peptidiques avec des affinités nanomolaires à micromolaires (μM) et neutraliser efficacement leur activité antimicrobienne ou hémolytique. Pour la protéine désordonnée NPM1, associée à la leucémie myéloïde aiguë, les peptides générés par le modèle ont montré une colocalisation nucléolaire dans les cellules vivantes, fournissant ainsi la première preuve in vivo de la capacité des protéines conçues par IA à se lier à des protéines naturellement désordonnées.
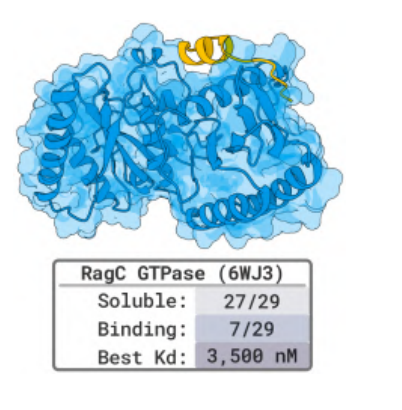
La conception de RagC, une enzyme essentielle du métabolisme cellulaire, et des dimères RagA:RagC a également donné des résultats remarquables :Sept des 29 peptides candidats se sont liés avec succès à RagC, l'affinité la plus élevée atteignant 3,5 μM ; 14 des conceptions de peptides à liaison disulfure cyclique ont montré une liaison stable.
BoltzGen a également démontré des capacités de conception à grande échelle sur deux petites molécules d’intérêt biomédical.Les liants protéiques obtenus ont montré une activité de liaison détectable dans la plage de 50 à 150 µM, démontrant que le modèle peut reconnaître de petites molécules sans intervention d'un chimiste expert. De plus, lors de la conception de peptides antimicrobiens ciblant l'ADN gyrase bactérienne GyrA, des séquences candidates supérieures à 19% ont réduit la croissance bactérienne de plus de quatre fois, certains peptides tuant directement les cellules hôtes.
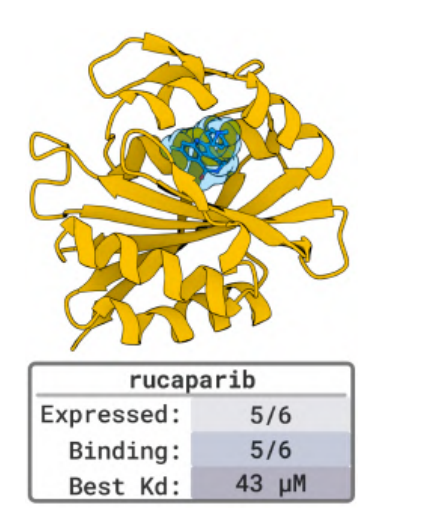
Dans les 5 tests cibles de référence avec des structures de liaison connues (telles que PD-L1, TNFα, PDGFR, etc.),BoltzGen a également atteint un taux de réussite élevé : des liants nanomolaires sont apparus sur la cible du 80%, vérifiant que sa précision est comparable à celle du meilleur modèle actuel.

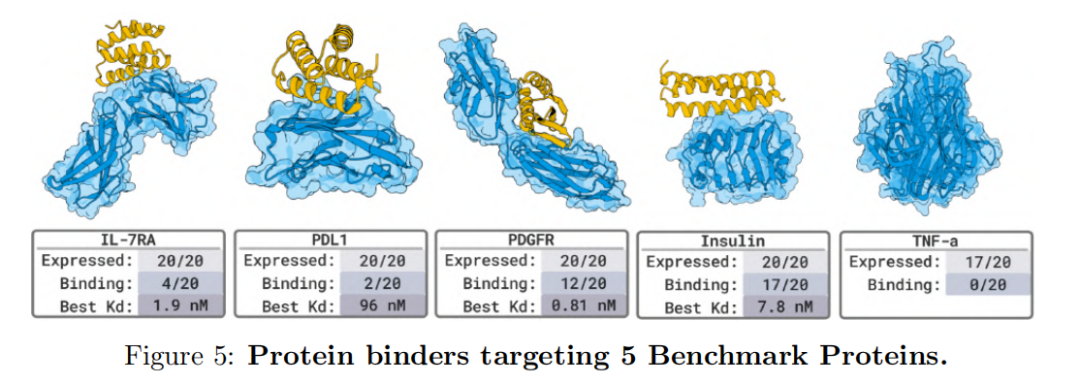
Globalement, cette série d'expériences démontre que BoltzGen peut non seulement reproduire des structures de liaison de haute qualité dans des distributions de données connues, mais aussi réaliser une conception fonctionnelle dans des systèmes biologiques totalement inconnus. Son architecture unifiée de génération tout-atome intègre le processus « conception-prédiction-vérification », offrant ainsi une infrastructure d'IA ouverte, contrôlable et évolutive pour la découverte de médicaments et l'ingénierie biomoléculaire.
De la prédiction à la génération, la série Boltz remodèle le paysage de la conception moléculaire pilotée par l'IA
En 2024,L’équipe de recherche de la clinique Jameel du MIT a présenté le modèle Boltz-1.Alors que l'industrie mondiale de la conception de médicaments passe de la prédiction de structure à la génération de fonctions, et que la série de modèles AlphaFold a été pionnière en matière de calculabilité du repliement des protéines, la disponibilité limitée d'AlphaFold3 restreint la capacité de l'industrie à itérer librement dans des scénarios médicamenteux réels. Boltz-1 est né dans ce contexte. Non seulement ses performances sont proches de celles d'AlphaFold3, mais il est également entièrement open source et commercialement viable, propulsant la prédiction de structure moléculaire dans l'écosystème ouvert de l'industrie.
Boltz-1 utilise un système de génération qui combine un modèle de diffusion avec une architecture de transformateur.Il permet de prédire les structures des protéines, de l'ARN, de l'ADN et des complexes de petites molécules à l'échelle atomique. Son interface conditionnelle flexible permet une modélisation précise de sites de liaison spécifiques ou de conformations moléculaires, élargissant ainsi considérablement ses applications industrielles. De la conception de nouveaux anticorps et de l'optimisation de l'ingénierie enzymatique au criblage de ligands de petites molécules, des prédictions de bout en bout peuvent être réalisées dans le cadre de Boltz-1, réduisant ainsi considérablement les obstacles à l'entrée en bioinformatique.
En 2025,L’équipe de la clinique Jameel du MIT a présenté le modèle Boltz-2 basé sur Boltz-1.Il a poussé la précision de la prédiction du repliement des protéines à un nouveau niveau et est connu sous le nom de « GPT-4 de la biologie structurale ».
Comparé à son prédécesseur, Boltz-2 améliore significativement la précision de génération et l'efficacité de calcul. Il introduit également une entrée conditionnelle multimodale, lui permettant d'intégrer les informations de séquence, les données expérimentales et les propriétés chimiques, permettant ainsi une conception moléculaire plus fine. Alors que le paysage mondial de la bioinformatique et de la découverte de médicaments évolue vers la génération de scénarios complets, Boltz-2 répond à la demande du monde universitaire et de l'industrie pour des outils hautement disponibles, évolutifs et commercialement viables.
Boltz-2 hérite et optimise le système de génération hybride du modèle de diffusion et de l'architecture Transformer.Son module principal Trunk peut extraire simultanément des représentations à plusieurs niveaux de complexes de protéines ou d'acides nucléiques.Le module Diffusion génère et optimise la structure sur cette base.
Grâce à une interface conditionnelle flexible, les chercheurs peuvent contrôler précisément la structure de sortie de sites de liaison spécifiques, de poches actives ou de ligands de petites molécules, ce qui élargit considérablement le potentiel d'application du modèle dans des domaines tels que la conception de nouveaux anticorps, l'optimisation de la catalyse enzymatique et le criblage de médicaments. Le caractère open source de Boltz-2 garantit également une itération libre entre le monde universitaire et l'industrie, accélérant ainsi l'application du calcul génératif moléculaire dans des scénarios concrets de développement de médicaments.
Aujourd'hui, BoltzGen a proposé un « langage de spécification de conception » universel qui permet au modèle de basculer de manière flexible entre différents systèmes tels que les protéines, les nanoanticorps, les peptides cycliques, les petites molécules, etc., pour réaliser une génération de structure intermodale et un contrôle des contraintes, élargissant encore le champ d'application de l'IA générative dans le domaine de la conception biomoléculaire.








