Command Palette
Search for a command to run...
RNA-Strukturvorhersage Konkurriert Mit AlphaFold 3! Ein Team Der Virginia Tech Stellt RNAbpFlow Vor, Das Völlig Unabhängig Von Evolutionären Informationen ist.

Im November 2020 erlangte AlphaFold 2 auf dem internationalen Wettbewerb CASP14 zur Vorhersage von Proteinstrukturen große Bekanntheit. Mit seiner nahezu experimentellen Genauigkeit bei der Vorhersage der dreidimensionalen Struktur von Proteinen löste AlphaFold 2 nicht nur das Problem der Proteinfaltung, das die biologische Forschungsgemeinschaft ein halbes Jahrhundert lang beschäftigt hatte, sondern verhalf seinen beiden Entwicklern vier Jahre später mit seinen bahnbrechenden Ergebnissen auch zum Nobelpreis für Chemie. Doch der Erfolg von AlphaFold 2 war erst der Anfang. Wie eine Atombombe entfachte es ein Feuerwerk in der gesamten Strukturbiologie und löste eine Welle von Forschungsarbeiten aus, die künstliche Intelligenz zur Analyse der räumlichen Konformation biologischer Makromoleküle nutzen.
Inspiriert von den bahnbrechenden Erfolgen von AlphaFold 2 auf dem Gebiet der Proteine, ruhen große Hoffnungen auch auf dem Einsatz künstlicher Intelligenz zur Lösung des Problems der Vorhersage der dreidimensionalen Struktur von Ribonukleinsäure (RNA).Allerdings stoßen die derzeitigen gängigen Algorithmen bei ihrer Implementierung immer noch auf mehrere unvermeidbare praktische Engpässe:Erstens stützen sich die meisten Transformer-basierten RNA-Vorhersagemodelle stark auf dominante evolutionäre Sequenzinformationen, die durch multiple Sequenzalignments (MSA) oder indirekt aus biologischen Sprachmodellen gewonnene homologe Sequenzinformationen bereitgestellt werden. Die isostrukturelle Natur der RNA-Basenpaarung erschwert jedoch die Generierung hochwertiger und zuverlässiger MSA-Ergebnisse. Zweitens erfassen die meisten gängigen Methoden die Schlüsselinformationen der Basenpaarung in der RNA-Sekundärstruktur nicht vollständig, obwohl diese den Kernfaktor für die dreidimensionale Faltungsmorphologie der RNA darstellt. Drittens besitzen RNA-Moleküle eine natürliche Konformationsflexibilität und existieren in mehreren stabilen räumlichen Konformationen. Bestehende Algorithmen liefern meist nur einzelne statische Ergebnisse und können die tatsächliche dynamische Konformationsmenge der RNA nicht rekonstruieren.
Um die zuvor genannten Herausforderungen zu bewältigen, arbeiteten Professor Debswapna Bhattacharya und sein Student Sumit Tarafder von Virginia Tech zusammen, um ein SE(3)-äquivariantes Flow-Matching-Modell, RNAbpFlow, zu entwickeln, das auf Sequenz- und Basenpaarbedingungen basiert.Dieses Modell kann allein auf Basis von RNA-Nukleotidsequenzen und Basenpaarungsinformationen einen vollständigen Satz von RNA-Konformationen aller Atome generieren, ohne dass evolutionäre Informationen wie multiple Sequenzalignments oder homologe Vorlagen erforderlich sind.Es behebt mehrere Schwächen bestehender KI-basierter Methoden zur RNA-Strukturvorhersage. Mehrere Benchmark-Experimente haben gezeigt, dass die Einbeziehung der Basenpaarung als Randbedingung die Vorhersagegenauigkeit des Modells signifikant verbessern kann. In den beiden Kernaufgaben der RNA-Topologie-Erfassung und der 3D-Konformationsmodellierung übertrifft RNAbpFlow gängige Standardmethoden.
Erwähnenswert ist, dass im Vergleichstest zwischen RNAbpFlow und AlphaFold 3 eine Reihe beeindruckender Daten das Endergebnis offenbarte:Im Blindtest-Experiment CASP16 war RNAbpFlow in der Lage, die globale Faltungstopologie der überwiegenden Mehrheit der 14 RNA-Zielsequenzen (≤200 nt) genau zu rekonstruieren, wobei 12 davon die Kriterien für eine qualifizierte Vorhersage erfüllten.Im Gegensatz dazu erfüllten unter den gleichen experimentellen Bedingungen nur 8 der vorhergesagten Zielstrukturen von AlphaFold 3 den Standard für die Übereinstimmung mit der natürlichen Konformation.
Die entsprechenden Forschungsergebnisse mit dem Titel „RNAbpFlow: base pair-augmented SE(3) flow matching for conditional RNA 3D structure generation“ wurden in Nature Methods veröffentlicht.
Forschungshighlights:
- Das vorgeschlagene RNAbpFlow ist ein SE(3) isofluidisches Matching-Modell, das auf Sequenz- und Basenpaarbedingungen basiert und mehrere Mängel bestehender Methoden zur Vorhersage der dreidimensionalen RNA-Struktur behebt.
- Es wurden drei wichtige Neuerungen vorgeschlagen: ein dreikanaliger, bedingter Input für die Basenpaarung, eine Charakterisierung des Nukleosidbasenzentrums sowie basenpaarungsspezifische Hilfsverluste (bp2D und bp3D), die die Genauigkeit der Basenpaarung und der dreidimensionalen Struktur deutlich verbesserten.
- Wir schlagen eine Strategie für unabhängige und nicht-repetitive Trainings- und Testdatensätze vor und verwenden mehrere maßgebliche Benchmarks sowie CASP-Blindtests zur Validierung. Die Gesamtleistung übertrifft die bestehender gängiger Algorithmen.

Papieradresse:
https://www.nature.com/articles/s41592-026-03128-4
Die unabhängige Deduplizierung von Trainings- und Testdatensätzen gewährleistet eine korrekte und faire Bewertung.
Um das selbstentwickelte Modell RNAbpFlow zu erstellen und die Authentizität und Zuverlässigkeit seiner Auswertungsergebnisse zu gewährleisten, verwendet diese Studie unabhängige Trainings- und Testdatensätze ohne inhaltliche Überschneidungen und weist verschiedenen Test-Benchmarks spezifische Modellgewichte zu, anstatt während des gesamten Prozesses ein einzelnes Standardmodell zu verwenden, um zu vermeiden, dass Trainingsdaten in den Auswertungsprozess einfließen und zu Ergebnisverzerrungen führen.
Hinsichtlich Modellentwicklung und interner Validierung,Diese Studie verwendet den RNA3DB-Datensatz. Dieser Datensatz eignet sich hervorragend zum Trainieren von Deep-Learning-Modellen und für interne Benchmarks, da er sowohl auf Sequenz- als auch auf Strukturebene keine Redundanz aufweist. In dieser Studie wurde die am 26. April 2024 aus der Proteindatenbank (PDB) extrahierte Version des RNA3DB-Datensatzes verwendet. Zur Auswahl repräsentativer RNA-Sequenzen für die Experimente wurde das im Originalartikel beschriebene Schema zur Aufteilung in Trainings- und Testdaten angewendet.
Um sicherzustellen, dass der Datensatz ausschließlich hochwertige natürliche RNA-Strukturen und präzise Basenpaarungsinformationen enthält, wurde das Experiment einer mehrstufigen Qualitätskontrolle unterzogen. Diese umfasste das Entfernen von Strukturen mit nur einem Atom pro Nukleotid sowie von Strukturen, die mit Proteinresten vermischt waren; das Kürzen kontinuierlicher experimenteller Sequenzen, um Fehlpaarungen zwischen FASTA-Sequenzen und dreidimensionalen Strukturen zu korrigieren und die Integrität der Basenpaarungen zu wahren; und das Löschen von Sequenzen ohne Basenpaarungen aus natürlichen Strukturen. Nach der Extraktion der Basenpaarungsinformationen mithilfe von RNAView, dem Herausfiltern kontinuierlicher ungepaarter RNA-Ketten mit einer Länge von ≥ 20 Nukleotiden und der Begrenzung der Sequenzlänge auf 30–200 Nukleotide wurden schließlich ein reiner Trainingsdatensatz mit 560 RNA-Sequenzen (Sequenzen, die nicht zur Rfam-Familie passen, wurden entfernt, um das Risiko von Datenlecks weiter zu minimieren) und ein reiner Testdatensatz mit 48 Sequenzen erhalten.
Erwähnenswert ist, dass dieser Teil der Datenpartitionierung vollständig der nicht-redundanten Aufteilung von RNA3DB folgt und es keine Überschneidungen in Sequenzen oder Strukturen zwischen den Trainings- und Testdatensätzen gibt, um ein Datenleck aus der Wurzel zu vermeiden.
Im Vergleichsexperiment von RNAbpFlow und RNAJP,Die Studie nutzte einen Datensatz von 22 RNA-Sequenzen mit Dreiweg-Konnektoren, ähnlich denen der RNAJP-Studie, und führte anschließend ein Screening durch. Die Forschenden entfernten zunächst Multimerstrukturen aus diesem Datensatz und verglichen die verbleibenden Sequenzen dann umgehend mit dem zuvor genannten bereinigten Trainingsdatensatz, um Redundanzen und Sequenzduplikationen zu eliminieren. Der endgültige Datensatz umfasste 12 Sequenzen.
Hinsichtlich der Kompatibilität mit den internationalen Wettbewerben CASP15 und CASP16,Um die Regeln des Blindtests im Wettbewerb strikt einzuhalten und einen fairen Vergleich mit anderen Algorithmen zu gewährleisten, wurde die von RNA3DB vorgegebene Aufteilung in Trainings- und Testdatensätze nicht direkt übernommen. Stattdessen wurden zwei vollständig disjunkte und sich nicht überschneidende neue Trainingsdatensätze aus RNA3DB erstellt und geplant.
Da die erste Charge der im CASP15-Wettbewerb zu testenden RNAs erst im Mai 2022 veröffentlicht wurde, berücksichtigten die Forscher bei der Erstellung des neuen Trainingsdatensatzes ausschließlich RNA-Sequenzen, die vor April 2022 in die PDB-Datenbank hochgeladen worden waren (alle aus RNA3DB). Dadurch wurde sichergestellt, dass die im CASP15 zu testenden RNAs beim Modelltraining vollständig vermieden wurden und die Regeln für das Blindtesting strikt eingehalten wurden. Der Trainingsdatensatz umfasste schließlich 731 RNA-Sequenzen mit einer Länge von 30 bis 784 Nukleotiden. Der für die Modellevaluierung verwendete Testdatensatz bestand aus 6 natürlichen und 4 synthetischen RNAs aus dem CASP15-Benchmark-Datensatz.
Für CASP16,Der im Experiment verwendete Trainingsdatensatz bestand aus 994 experimentell aufgelösten PDB-Strukturen und 2.170 hochkonfidenten Vorhersagestrukturen, die zur Datenerweiterung mittels Kreuzdestillation verwendet wurden, insgesamt also aus 3.164 Proben.Das Verhältnis der Anzahl der PDB-Strukturen zur Anzahl der kreuzdestillierten Daten in jedem Trainingsbatch beträgt ungefähr 1:2,2.
Aus demselben Grund wurde die erste Charge der in CASP16 zu testenden RNAs im Mai 2024 veröffentlicht. Daher fassten die Forscher alle geeigneten Daten aus RNA3DB zu einem einzigen Trainingsdatensatz zusammen, der ausschließlich PDB-Strukturen enthielt, die bis zum 6. April 2024 hochgeladen worden waren. Nach einem strengen Screening wurden 994 RNA-Sequenzen und ihre entsprechenden gemessenen dreidimensionalen Strukturen ermittelt. Die Testdaten stammten von 28 RNAs, deren experimentelle dreidimensionale Strukturen aktuell in CASP16 verfügbar sind.
Die zur Erstellung des Kreuzdestillationsdatensatzes verwendeten Daten stammen aus dem bpRNA-1m(90)-Datensatz, vorwiegend um die Auswirkungen der Datenaugmentation zu untersuchen. Der bpRNA-1m(90)-Datensatz enthält 28.370 RNA-Sequenzen (und ihre entsprechenden Sekundärstrukturen). Nach dem Entfernen von Redundanzen, der Begrenzung der Sequenzlänge auf 30–200 Aminosäuren und nach mehrfacher Extraktion mithilfe des Clustering-Tools MMseqs2,Das Endergebnis war ein Kreuzdestillationsdatensatz von 2.170 RNA-Strukturen mit hoher Konfidenz.
Framework für bedingte Generierung basierend auf Stream-Matching
RNAbpFlow ist ein SE(3)-isofluidisches Matching-Modell, das auf Sequenz- und Basenpaarbedingungen basiert (siehe Abbildung unten). Es ist eine Modifikation des Proteinstrukturgenerierungsmodells FrameFlow und verwendet die von NuFold vorgeschlagene Nukleotid-Repräsentationsmethode, die jedes Nukleotid der RNA-Sequenz als starren Rahmen darstellt. Unter Verwendung von Nukleotidsequenz- und Basenpaarungsinformationen als Randbedingungen sagt es Diederwinkel durch iteratives Sampling voraus und reduziert so schrittweise alle Atome auf ihre endgültige, vollständige dreidimensionale Struktur der RNA mit der wahren Ribose-Faltungskonformation. Kurz gesagt,Die wichtigsten Vorteile von RNAbpFlow lassen sich in zwei Punkten zusammenfassen: „Eingangsbedingungen“ und „Modellparadigma“.
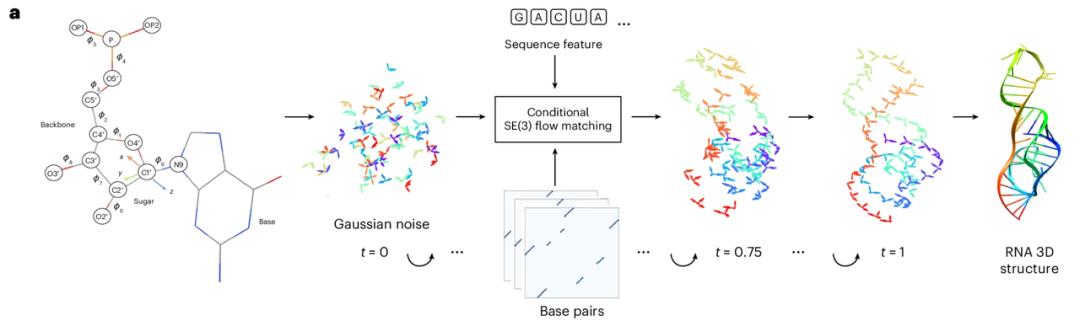
Bezüglich der "Eingangsbedingungen",RNAbpFlow kann bedingt dreidimensionale RNA-Strukturen erzeugen, ohne dass MSA- oder homologe Template-Informationen erforderlich sind; es stützt sich ausschließlich auf Sequenz- und Basenpaarungsinformationen.
Zunächst wird für die Sequenz eine RNA-Sequenz der Länge L eingegeben, wobei die Nukleotide mittels One-Hot-Encoding dargestellt werden. Das heißt, ein Binärvektor mit vier Elementen entspricht den vier Nukleotidtypen (A, U, C und G). Anschließend werden für die Verarbeitung der Basenpaarungsinformationen während der Trainingsphase (die zentrale Innovation) drei Tools – RNAView, MC-Annotate und DSSR – verwendet, um zweidimensionale Paarungsannotationen aus der experimentell ermittelten natürlichen dreidimensionalen Struktur zu extrahieren. Diese werden als drei unabhängige L × L Binärmatrizen dargestellt. Um verschiedene klassische und nicht-klassische Basenpaarungsmerkmale vollständig zu erfassen, werden die von den drei Tools erhaltenen widersprüchlichen Annotationen nicht einheitlich korrigiert. Stattdessen werden die drei Binärmatrizen direkt zu einem L × L × 3 Tensor verkettet, der als Eingabe für den Bias-Term in der Entrauschungsnetzwerkstruktur dient und Paarungsmerkmalsinformationen in drei unabhängigen Kanälen bereitstellt.
Da für die Sampling- und Inferenzszenarien der Testziele der Wettbewerbe CASP15 und CASP16 keine exakten Werte für natürliche Basenpaarungen vorliegen, wurden in den Experimenten sequenzabhängige Paarungsvorhersagematrizen verwendet, die von drei RNA-2D-Strukturvorhersage-Tools ausgegeben wurden: IPKnot, SPOT-RNA und RibonanzaNet. Alle drei Tools unterstützen die Basenpaarungsvorhersage basierend auf der Identifizierung von Pseudoknoten. Ihre Auswahl basierte auf ihrer Sampling-Leistung bei natürlichen RNA-Zielen in CASP15. Darüber hinaus ist RNAbpFlow sehr vielseitig und ermöglicht es Benutzern, drei Sätze von Paarungsmatrizen als Eingabe zu definieren. Falls nur ein Satz benutzerdefinierter Paarungsmatrizen verfügbar ist, kann dieser dreimal kopiert werden, um dem vom Netzwerk benötigten dreikanaligen 2D-Eingabeformat zu entsprechen.
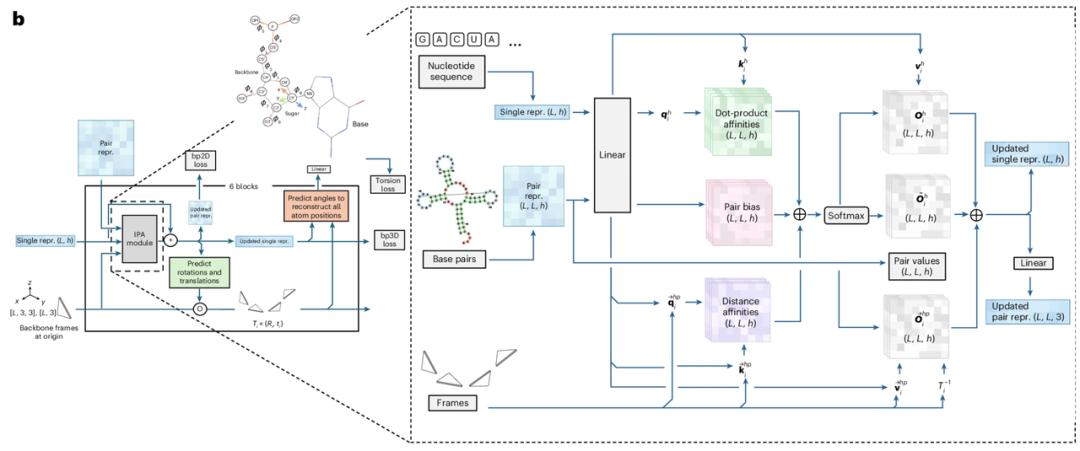
Im Hinblick auf das „Modellparadigma“,Der Schlüssel liegt im „Flow Matching“. Flow Matching ist ein Typ von tiefen generativen Modellen, dessen Hauptziel darin besteht, ein Geschwindigkeitsfeld (oder Flussfeld) zu lernen, das dem Wahrscheinlichkeitsfluss der Datenverteilung entspricht. Dadurch werden einfache Verteilungen wie Gaußverteilungen in die komplexe Datenverteilung transformiert, die für das Ziel in einem hochdimensionalen Raum erforderlich ist. Flow Matching lernt dieses Geschwindigkeitsfeld direkt, um die Bewegung von Stichprobenpunkten zu beschreiben, die von einer einfachen Verteilung zur Zielverteilung wandern, ohne die ursprüngliche Datenverteilung vollständig zu verändern. Durch die Integration gewöhnlicher Differentialgleichungen auf das gelernte Vektorfeld kann Flow Matching einfachere Migrationstrajektorien generieren, um das Ziel zu approximieren. Im Vergleich zu Diffusionsmodellen kann Flow Matching die Rechengeschwindigkeit bei der Generierung großer Stichproben deutlich verbessern.
Ziel dieser Studie ist es, mithilfe der Flow-Matching-Methode ein parametrisiertes Vektorfeld Ut zu erlernen. Dieses Vektorfeld repräsentiert eine stetige, zeitlich veränderliche Abbildung, aus der gewöhnliche Differentialgleichungen abgeleitet werden, um die Transformationsbeziehung zwischen zwei Verteilungen zu beschreiben: der Verteilung p₀(T₀) (verrauschte Frames) und der Verteilung p₁(T₁) (wahre Referenzframes). Um diese Abbildung zu erlernen, trainierten die Forscher ein parametrisiertes neuronales Netzwerk Vθ(Tₜ, t), das das Vektorfeld basierend auf dem verrauschten wahren Referenzframe Tₜ zum Zeitpunkt t vorhersagt. Dieser Teil des Netzwerks wurde in Anlehnung an FrameFlow entworfen und verwendet Strukturmodule von AlphaFold 2 als Basisarchitektur.
Hinsichtlich der spezifischen Trainingseinstellungen wurde das Experiment mit dem PyTorch-Lightning-Framework durchgeführt, um das Modell zu trainieren. Dabei wurde der Adam-Optimierer mit einer Lernrate von 0,0001 verwendet. Der verteilte Trainingsprozess lief auf 8 NVIDIA H100 GPUs über 1500 Iterationen.
Die missionskritische Leistung übertrifft die von AlphaFold 3
Zur Bewertung der Sampling-Leistung von RNAbpFlow wurde es zunächst mit RNAJP verglichen, einer dreidimensionalen RNA-Struktur-Sampling-Methode, die auf grobkörnigen Molekulardynamiksimulationen basiert und Basenpaarungsinformationen explizit berücksichtigt, einschließlich nicht-klassischer Basenpaarung, Basenstapelungsinteraktionen und Schleifen-Schleifen-Wechselwirkungen über große Entfernungen.
Die experimentellen Ergebnisse sind in der folgenden Abbildung dargestellt.RNAbpFlow schnitt in beiden Bewertungskriterien besser ab als RNAJP.RNAbpFlow erzielte insbesondere einen hohen durchschnittlichen Wert von 0,66 im lokalen Distanzdifferenztest (lDDT-Wert), während RNAJP nur 0,59 erreichte. Auch beim globalen Topologie-Sampling erzielte RNAbpFlow einen durchschnittlichen Template-Modellierungs-Wert (TM-Wert) von 0,38, während RNAJP 0,32 erreichte.
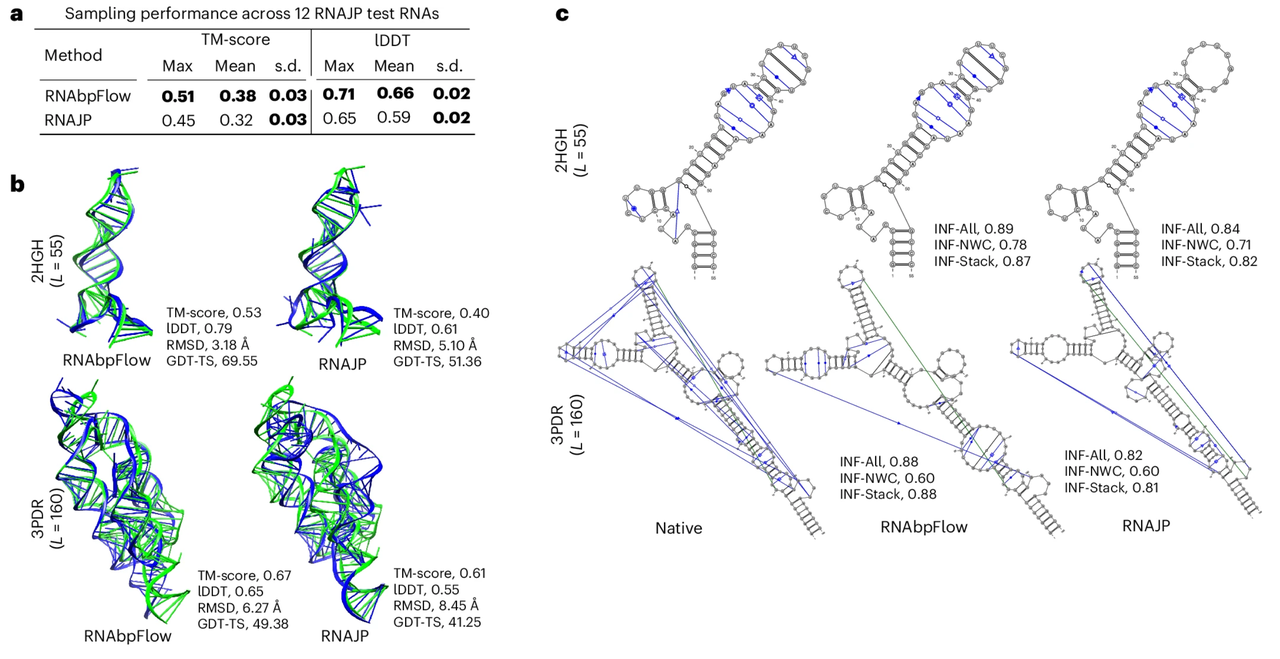
Hinsichtlich der Bewertung der Gültigkeit der Stichproben wird die korrekte Faltstruktur anhand des TM-Scores bestimmt.RNAbpFlow detektierte RNA-Ziele bis zu 66.671 TP3T.Die Erkennungsrate korrekt gefalteter Strukturen basierend auf lDDT-Scores betrug 251 TP3T; im Gegensatz dazu lagen die Raten für RNAJP bei lediglich 41,671 TP3T bzw. 0 TP3T. Von den 12.000 mit RNAbpFlow generierten simulierten Konformationen wiesen 13,41 TP3T-Konformationen Template-Modellierungs-Scores über 0,45 und 9,61 TP3T-Konformationen lDDT-Scores über 0,7 auf. Im Gegensatz dazu erreichten von den simulierten RNAJP-Konformationen nur 1,731 TP3T Template-Modellierungs-Scores über 0,45, und keine Konformation einen lDDT-Score über 0,75.
Die obigen Ergebnisse zeigen, dass RNAbpFlow RNAJP nicht nur bei der Bewertung optimaler Strukturen übertrifft, sondern auch einen höheren Prozentsatz an qualitativ hochwertigen simulierten Konformationen erreicht.Dies unterstreicht seine Effizienz bei Dual-Sampling-Aufgaben, die sowohl die globale Topologie als auch die lokale Konfiguration betreffen.
Anschließend verglich das Forschungsteam RNA bpFlow mit mehreren bestehenden Methoden anhand des CASP15-Blindtestdatensatzes (siehe Tabelle unten). Die Ergebnisse zeigen, dass…Bei Eingabe realer und genauer Informationen über natürliche Basenpaarungen wird die Modellierungs- und Vorhersageleistung von RNAbpFlow deutlich verbessert.Der durchschnittliche TM-Score erreichte 0,48, die mittlere quadratische Abweichung aller Atome (RMSD) betrug 7,77 und die Genauigkeit des Nicht-Watson-Crick-Basenpaar-Interaktionsnetzwerks (INF-NWC) betrug 0,62; wenn nur die vom Algorithmus vorhergesagten Basenpaarungen verwendet wurden, betrugen die drei Indizes 0,40, 10,70 bzw. 0,48.
Im Vergleich dazu erhöhte sich bei Bereitstellung von Informationen über die korrekte Basenpaarung der TM-Score um 201 TP3T, der RMSD-Wert sank um 27,41 TP3T und der INF-NWC-Wert stieg um 29,21 TP3T, was die Bedeutung qualitativ hochwertiger Basenpaarungsinformationen unterstreicht.
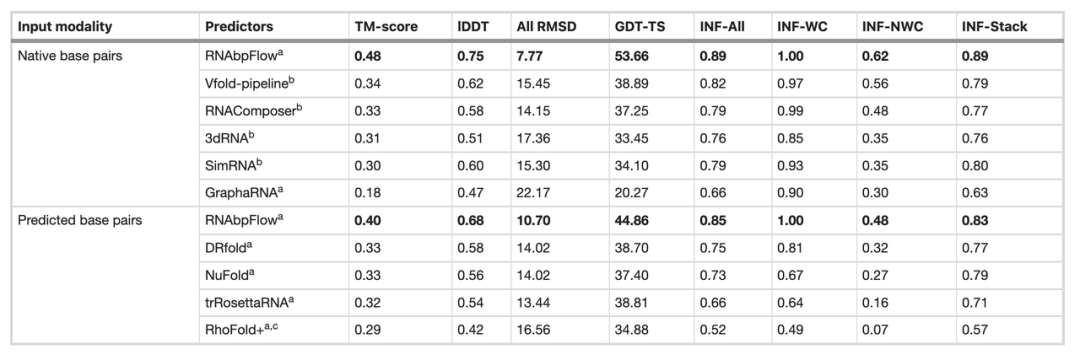
Im Gegensatz dazu zeigen andere Methoden selbst bei der Eingabe realer natürlicher Basenpaare nur eine sehr geringe Leistungsverbesserung. Beispielsweise erreicht Vfold, die leistungsstärkste Methode, lediglich einen TM-Score von 0,34, während RNAComposer einen minimalen RMSD-Wert von 14,15 aufweist. Dies belegt, dass RNAbpFlow eine höhere Anpassungsfähigkeit besitzt, präzise Basenpaarungsbeschränkungen in tiefen generativen Modellen effizient nutzt und die obere Grenze der Vorhersageleistung von KI-Methoden deutlich anhebt.
Um die Genauigkeit der Probenentnahme von RNAbpFlow anhand der Blindtest-Targets von CASP16 zu überprüfen, verglichen die Forscher es mit den beiden leistungsstärksten Vorhersagemodellen des CASP16-Wettbewerbs. Die Ergebnisse (siehe Abbildung unten) zeigen, dass RNAbpFlow alle Algorithmen, einschließlich AlphaFold 3, hinsichtlich des durchschnittlichen maximalen TM-Scores und der lDDT-Metriken für 14 vorhergesagte Targets (≤ 200 nt) übertrifft. Von den von RNAbpFlow generierten Strukturkonformationen konnte in 12 (85.711 TP3T) mindestens eine korrekt gefaltete Konformation gefunden werden; AlphaFold 3 erreichte dies hingegen nur in 8 (57.131 TP3T) Konformationen, was die stabilere Leistung von RNAbpFlow belegt.
Bei RNA-Zielsequenzen mit mehr als 200 Nukleotiden schneidet RNAbpFlow zwar immer noch besser ab als NuFold, trRosettaRNA2 und DRfold2, aber schlechter als AlphaFold3. Dies liegt daran, dass die Genauigkeit der vorhergesagten Basenpaarungen bei langen Sequenzen deutlich abnimmt.
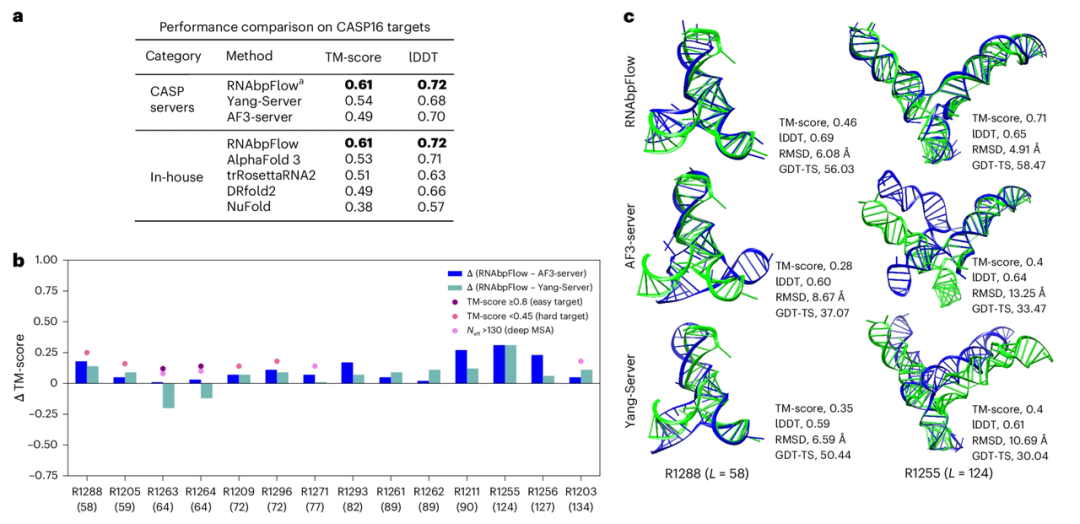
Die Experimente zeigten zudem, dass das auf Basenpaarungsbeschränkungen basierende Strukturvorhersagemodell bei unvorhersagbaren Zielsequenzen (TM-Score < 0,45; effektive Homologiesequenztiefe der MSA ≤ 130; schwaches evolutionäres Signal) RNAbpFlow deutlich übertraf, insbesondere bei geringer Verfügbarkeit evolutionärer Informationen. Dies unterstreicht die Überlegenheit von RNAbpFlow, das die dreidimensionale Struktur von RNA allein anhand von Basenpaarungsinformationen vorhersagen kann. Bei leicht vorhersagbaren Zielsequenzen wie R1263 und R1264 hingegen ermöglichten ausreichend tiefe multiple Sequenzalignment-Daten den beiden Alignment-Modellen AF3-Server und Yang-Server, mit RNAbpFlow gleichzuziehen oder es sogar zu übertreffen. Dies verdeutlicht die starke Abhängigkeit beider Modelle von Sequenzalignment-Informationen.
Letzte Worte
Zusammenfassend lässt sich sagen, dass RNAbpFlow nicht durch MSA und strukturelle Homologie eingeschränkt ist. Es kann direkt vollständige dreidimensionale RNA-Strukturmodelle von Ende zu Ende allein anhand von Sequenz und Basenpaarung generieren. Mithilfe hochpräziser Technologien zur Generierung großer Konformationssätze auf atomarer Ebene eröffnet es möglicherweise einen vielversprechenden neuen Weg für die Erforschung der RNA-Konformationsdynamik.








