Command Palette
Search for a command to run...
In Nur 30 Minuten Integrierte Das Biologische Multiagentensystem Robin Erfolgreich 550 Forschungsarbeiten, Etablierte Einen Autonomen Forschungskreislauf Und Identifizierte Kandidaten Für Therapien Gegen dAMD.

Mit der kontinuierlichen Weiterentwicklung biologischer Nachweismethoden, Perturbationsexperimente und computergestützter Modellierungstechnologien verbessern sich Präzision und Umfang der lebenswissenschaftlichen Forschung rasant. Im Vergleich zur schnell wachsenden Datenproduktionskapazität hinken die intelligenten Fähigkeiten des Forschungssystems in Bezug auf Wissensintegration und wissenschaftliches Denken jedoch deutlich hinterher.Eine Fülle wertvoller Informationen ist über wissenschaftliche Artikel, Datenbanken und experimentelle Ergebnisse verstreut. Die manuelle Sortierung ist nicht nur ineffizient, sondern erschwert auch die Verknüpfung bestehender Erkenntnisse aus verschiedenen Fachgebieten.Dies hat zur Folge, dass viele validierte Erkenntnisse nicht zeitnah in neue Forschungsideen oder klinische Protokolle umgesetzt werden konnten.
Dieses Problem der „Wissensfragmentierung“ tritt besonders deutlich im Bereich des „Arzneimittel-Repurposing“ zutage. Ob es sich um die spätere Entdeckung der otoprotektiven Wirkung von Dabrafenib oder die Erweiterung des neuen therapeutischen Nutzens von Ketamin handelte, beides führte zu Verzögerungen bei der Translation von mehreren Jahren oder sogar Jahrzehnten, was die aktuellen Engpässe bei der Wissensgewinnung und -integration im wissenschaftlichen Forschungsprozess widerspiegelt.
In den letzten Jahren haben große Sprachmodelle (LLMs) mit ihren durch das Training mit massiven Korpora entwickelten Fähigkeiten zum Abrufen, Induktivisieren und logischen Denken begonnen, ihr Potenzial in der lebenswissenschaftlichen Forschung unter Beweis zu stellen. Durch die Kombination von Feinabstimmung, abrufbasierter Generierung (RAG) und Multiagenten-Kollaborationstechniken haben diese Modelle die menschliche Leistung bei einzelnen Aufgaben wie Literaturanalyse, Wirkstoffvorhersage und Generierung wissenschaftlicher Hypothesen erreicht oder sogar übertroffen.Die meisten bestehenden KI-Tools decken immer noch nur bestimmte Teile des Forschungsprozesses ab und können die gesamte Kette von „Hypothesengenerierung – Versuchsplanung – Datenanalyse – Ergebnisiteration“ nicht wirklich miteinander verbinden.Daher kann eine wirklich geschlossene, intelligente wissenschaftliche Forschung noch nicht erreicht werden.
Um dieses Problem anzugehen, entwickelte ein gemeinsames Team von FutureHouse in San Francisco, der Universität Oxford und der Fordham University das biologische Multiagentensystem Robin.Dies ist das erste biomedizinische intelligente System, das gleichzeitig die Fähigkeit zur Generierung wissenschaftlicher Hypothesen und zur Analyse experimenteller Daten integriert und einen kontinuierlichen geschlossenen Arbeitsablauf erreicht.
Robin kann durch die Zusammenarbeit eines Literaturrecherche- und eines Datenanalyse-Tools Krankheitsmechanismen analysieren, Wirkstoffkandidaten screenen, experimentelle Studien auswerten und Hypothesen iterativ weiterentwickeln. Das Forschungsteam nutzte die trockene altersbedingte Makuladegeneration (dAMD), eine Erkrankung mit begrenzten Behandlungsmöglichkeiten und dringendem klinischem Bedarf, als Anwendungsbeispiel, um Robins Fähigkeiten im intelligenten Wirkstoff-Screening zu validieren. Dies liefert ein neues praktisches Paradigma für die KI-gestützte Entwicklung neuer Medikamente und das Drug Repurposing.
Die entsprechenden Forschungsergebnisse mit dem Titel „Ein Multiagentensystem zur Automatisierung wissenschaftlicher Entdeckungen“ wurden in Nature veröffentlicht.
Forschungshighlights:
* Das Robin-System ist das erste, das die Generierung von Literaturhypothesen und die Analyse biologischer experimenteller Daten in einen kontinuierlichen, geschlossenen Arbeitsablauf integriert.
Robin ist flexibel und kann auf multidisziplinäre Forschungsergebnisse zurückgreifen. Im Bereich der Arzneimittelentwicklung genügt die Eingabe des Namens der Zielkrankheit, und das System analysiert automatisch die wichtigsten pathologischen Mechanismen, gleicht sie mit In-vitro-Modellen ab, schlägt Wirkstoffkandidaten vor, führt die experimentelle Datenanalyse durch und optimiert die Kandidatenmoleküle iterativ.
Robin nutzte die diffuse altersbedingte Makuladegeneration als Forschungsbeispiel und schlug als Erster eine neuartige Strategie zur Behandlung der trockenen Makuladegeneration vor, indem er ROCK-Inhibitoren einsetzte, um die phagozytische Funktion des retinalen Pigmentepithels zu verbessern.

Lesen Sie das Dokument:
https://www.nature.com/articles/s41586-026-10652-y
Datensätze: Umfasst öffentliche Literatur, Bioinformatik-Benchmarks und experimentelle Daten.
In dieser Studie wurde ein dreistufiges Datensystem entwickelt, das aus öffentlich zugänglichen Literaturdaten, allgemeinen bioinformatischen Benchmark-Daten und selbstexperimentellen Daten besteht.Es umfasst verschiedene Aufgabentypen, darunter Literaturtexte, bioinformatische Analyseaufgaben, Zellerkennung und Transkriptomsequenzierung.Es deckt im Wesentlichen die wichtigsten Datenszenarien im KI-gestützten Arzneimittelentwicklungsprozess ab.
Zunächst integrierten die Forscher 551 chinesische und englische wissenschaftliche Forschungsarbeiten zum Thema dAMD als Wissensbasis für das System, um wissenschaftliche Hypothesen zu generieren.Dies umfasst 151 Studien zu Krankheitsmechanismen und 400 Forschungsarbeiten zur phagozytischen Funktion von retinalen Pigmentepithelzellen und deren Zusammenhang mit Krankheiten.Diese Literatur dient nicht nur der Aufklärung von Krankheitsmechanismen, sondern liefert auch eine theoretische Grundlage für das Screening von In-vitro-Versuchsmodellen und die Entwicklung von Wirkstoffkandidaten für die Wiederverwendung von Medikamenten. Sie bildet die zentrale Quelle für Robins Wissensgewinnungsbemühungen.
Zweitens verwendeten die Forscher den allgemeinen Bioinformatik-Benchmark-Datensatz BixBench, um die Datenanalysefähigkeiten des Systems quantitativ zu bewerten.Die Studie wählte 170 Testfragen zum Thema Arzneimittelentwicklung aus.Es umfasst verschiedene Aufgabentypen, darunter Transkriptomanalyse, Genomik, funktionelle Anreicherungsanalyse, Sequenzanalyse und statistische Tests. Alle Fragen beinhalten standardisierte Datenpakete, Standardantworten und Ablenkungsoptionen, mit denen sich die Anpassungsfähigkeit und Stabilität von Agenten in realen Bioinformatik-Szenarien systematisch bewerten lässt.
Auch,Die Forscher erstellten außerdem einen eigenen experimentellen Datensatz, um die Modelliteration und die experimentelle Überprüfung durch reale Daten zu unterstützen.Die Daten umfassen Durchflusszytometrie-Ergebnisse von ARPE-19-Zellen und humanen primären retinalen Pigmentepithel-Stammzellen, RNA-Seq-Transkriptomdaten nach verschiedenen Medikamentenbehandlungen sowie Ergebnisse von Zytotoxizitäts-, immunzytochemischen Färbungs- und VEGF-ELISA-Tests. Die humanen Zellproben stammen von der New York Vision Repair Eye Bank und waren ausschließlich retinale Pigmentepithel-Stammzellen von Spendern ab 60 Jahren ohne Augenerkrankungen. Dies gewährleistet die Authentizität und den klinischen Referenzwert der experimentellen Daten.
Robin: Multiagentensysteme für die biomedizinische Wissenschaftsforschung
Robin, basierend auf dem Aviary-Framework und ausgeführt in der Jupyter Notebook-Umgebung, unterscheidet sich von herkömmlichen KI-Forschungswerkzeugen, die nur einzelne Aufgaben ausführen. Es ist das erste, das einen kontinuierlichen, geschlossenen Workflow von „wissenschaftlicher Hypothesengenerierung – experimenteller Analyse – Ergebnisrückmeldung – Hypotheseniteration“ erreicht.Es kann den gesamten wissenschaftlichen Forschungsprozess, einschließlich der Erforschung von Krankheitsmechanismen, des Screenings von Wirkstoffkandidaten und der Analyse experimenteller Daten, halbautonom durchführen.
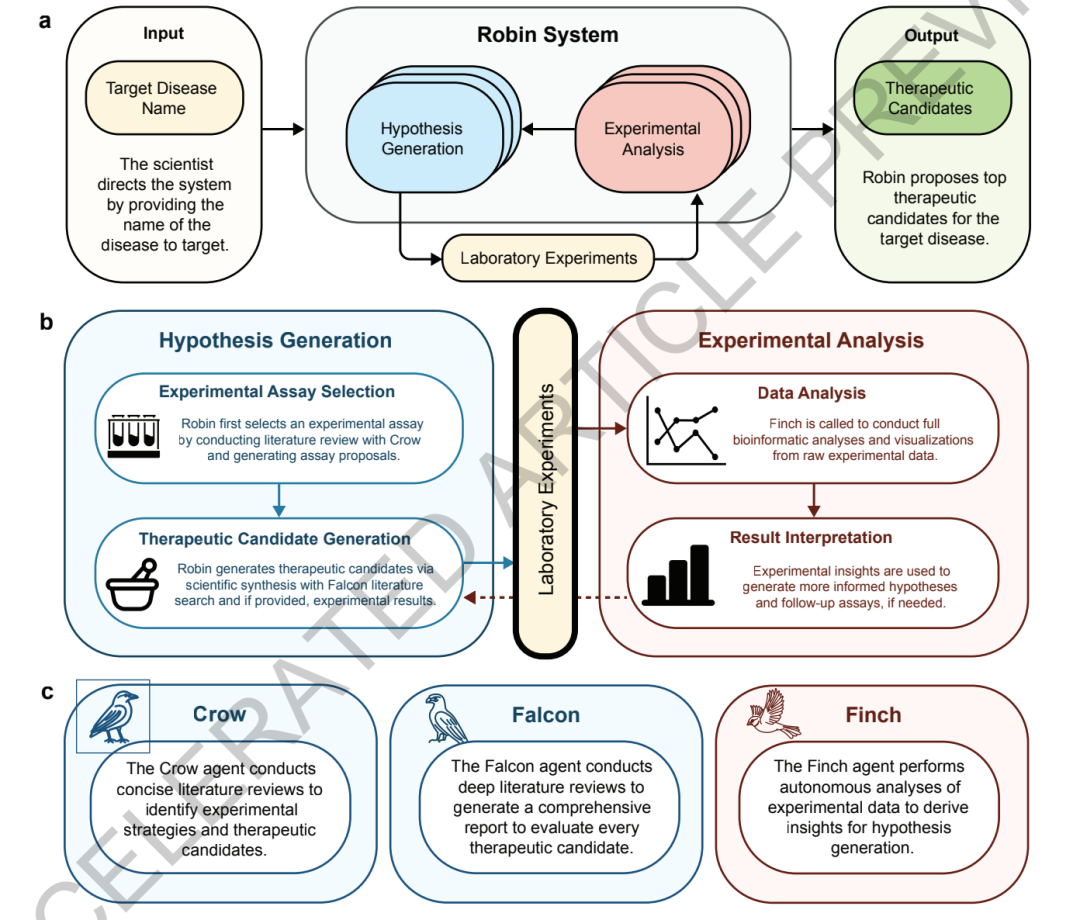
Das System verwendet eine „Drei-Agenten“-Kernarchitektur, bestehend aus zwei Dokumentenagenten und einem Datenanalyseagenten, die zusammenarbeiten.
In,Die beiden Dokumentenintelligenzagenten Crow und Falcon sind in erster Linie für die Gewinnung von Dokumentenwissen und die Generierung wissenschaftlicher Hypothesen verantwortlich.Beide Modelle basieren auf dem OpenAI o4-mini-Modell. Crow ist zuständig für die Recherche krankheitsbezogener Literatur, die Analyse pathologischer Mechanismen, das Screening experimenteller Modelle und die Identifizierung potenzieller Wirkstoffkandidaten. Es kann fragmentierte Forschungsergebnisse systematisch integrieren und wichtige wissenschaftliche Schlussfolgerungen extrahieren. Falcon übernimmt die detaillierte Validierung und Optimierung. Darüber hinaus analysiert es die pharmakologischen Mechanismen, die theoretischen Grundlagen und die potenziellen Einschränkungen der von Crow vorgeschlagenen Lösungsansätze und korrigiert fehlerhafte Zitate in der Literatur. Dadurch wird das Problem der „Illusion“ großer Modelle reduziert.
Das dritte Kernmodul, Finch, ist ein intelligenter Agent, der speziell für die Analyse biologischer experimenteller Daten entwickelt wurde.Im Gegensatz zu herkömmlichen Tools, die auf festen Analyseskripten basieren, nutzt Finch generative Inferenz. Basierend auf den Merkmalen der experimentellen Daten generiert und führt es Python- oder R-Code in Echtzeit aus. Dadurch kann es Aufgaben wie Durchflusszytometrie-Analysen, RNA-Seq-Differenzialexpressionsanalysen und Genfunktionsanreicherungen adaptiv durchführen. Das System ist somit nicht mehr auf vordefinierte Analyse-Workflows beschränkt, sondern kann seine Analysestrategien dynamisch anpassen – ähnlich wie ein Forscher.
Um die Zufälligkeit großer Modelle in der Datenanalyse zu reduzieren,Robin entwarf außerdem einen Mechanismus zur „Multi-Trajektorien-Analyse + Konsensintegration“.Das System kann gleichzeitig acht unabhängige Finch-Analyse-Trajektorien starten. Jede Trajektorie führt unabhängig Codegenerierung, Datenanalyse und Ergebnisausgabe durch. Abschließend werden die Ergebnisse mehrerer Trajektorien mittels Metaanalyse integriert. Dadurch werden Abweichungen, die durch Schwankungen und Parameterunterschiede in der Einzelanalyse entstehen, reduziert und die Stabilität der Ergebnisse verbessert.
Im Hinblick auf die Evaluierungsmechanismen führte Robin außerdem ein zweistufiges System zur Überprüfung groß angelegter Modelle ein.Das System verwendet Anthropics Claude 3.7 Sonnet als zentrales Bewertungsmodell und kombiniert es mit Google Gemini 2.5 Pro, um den Präferenzen von Fachexperten gerecht zu werden.Kandidatenmechanismen, experimentelle Modelle und Medikamentenregime werden hierarchisch mittels paarweiser Vergleiche und Turnierranking evaluiert. Bei einer geringen Anzahl zu evaluierender Regime wird ein vollständiger Paarvergleich durchgeführt; bei einer großen Anzahl erfolgt der Vergleich per Zufallsstichprobe. Das Bradley-Terry-Luce-Modell dient der Erstellung des gewichteten Rankings und gewährleistet so die Genauigkeit der Evaluierung bei gleichzeitiger Kontrolle des Rechenaufwands.
Um die Reproduzierbarkeit des Analyseprozesses zu gewährleisten, laufen alle Finch-Tasks in unabhängigen Docker-Containerumgebungen und sind mit einer vollständigen Bioinformatik-Toolchain vorinstalliert. Das Forschungsteam optimierte und vereinfachte den Workflow zudem durch mehrere Prompt-Engineering-Runden und komprimierte den komplexen ursprünglichen Prozess in einen stabilen und benutzerfreundlichen Jupyter-Workflow. Dies verbessert die Anwendbarkeit des Systems in Forschungsszenarien zusätzlich.
Robin entdeckte, dass Lipasudil die Phagozytosefähigkeit um das 1,89-fache erhöht.
Diese Studie konzentriert sich auf dAMD als zentrales Anwendungsszenario und entwirft mehrere Validierungsexperimente, die auf Robins Fähigkeiten zur Hypothesengenerierung, Datenanalyse, architektonischen Effektivität und der Effizienz der realen Arzneimittelentwicklung basieren.
Die Kernexperimente konzentrieren sich auf das Screening von Wirkstoffkandidaten und die Überprüfung des Wirkmechanismus. Robin identifizierte zunächst durch Literaturanalysen 10 wichtige pathogene Mechanismen der dAMD und bestimmte die „Verbesserung der phagozytischen Funktion der retinalen Pigmentepithelzellen“ als zentrale Behandlungsrichtung.In der ersten Screening-Runde schlug das System 30 Wirkstoffkandidaten vor. Die Forscher wählten Exenatid, Fingolimod, Y-27632 und weitere Wirkstoffe für experimentelle Untersuchungen aus und verwendeten das bekannte, wirksame Medikament MFGE8 als Positivkontrolle.
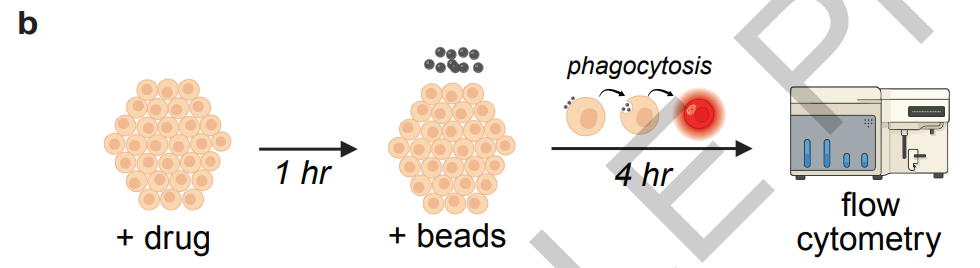
Anschließend entwickelte Robin unabhängig ein experimentelles Protokoll zur Transkriptomsequenzierung, und Finch führte die Datenanalyse durch. Die Ergebnisse zeigten, dass Y-27632 durch die Regulation des Aktin-Zytoskeletts, des Autophagie-Signalwegs und des Schlüsselgens ABCA1 für den Lipidtransport eine Transkriptom-Reprogrammierung von retinalen Pigmentepithelzellen bewirken kann, wodurch ein bisher unbekannter Wirkmechanismus aufgedeckt wurde.
Um die klinische Relevanz des Wirkstoff-Screenings weiter zu verbessern, wurde im Anschluss eine zweite Runde von Wirkstoff-Iterationsexperimenten durchgeführt. Robin fügte 10 neue Wirkstoffkandidaten hinzu und stellte fest, dass das auf dem Markt befindliche Glaukommedikament Ribasudil wirksamer war als Y-27632 und die zelluläre Phagozytosekapazität um etwa das 1,89-fache erhöhte.Das Forschungsteam verwendete anschließend humane primäre retinale Pigmentepithel-Stammzellen, die dem physiologischen Milieu näherkommen, für ein erneutes Screening. Die Ergebnisse bestätigten abermals die dosisabhängigen Effekte von Ribasudil und Y-27632 und zeigten, dass Ribasudil keine offensichtliche Zytotoxizität aufweist und ein hohes Potenzial für die klinische Anwendung besitzt.
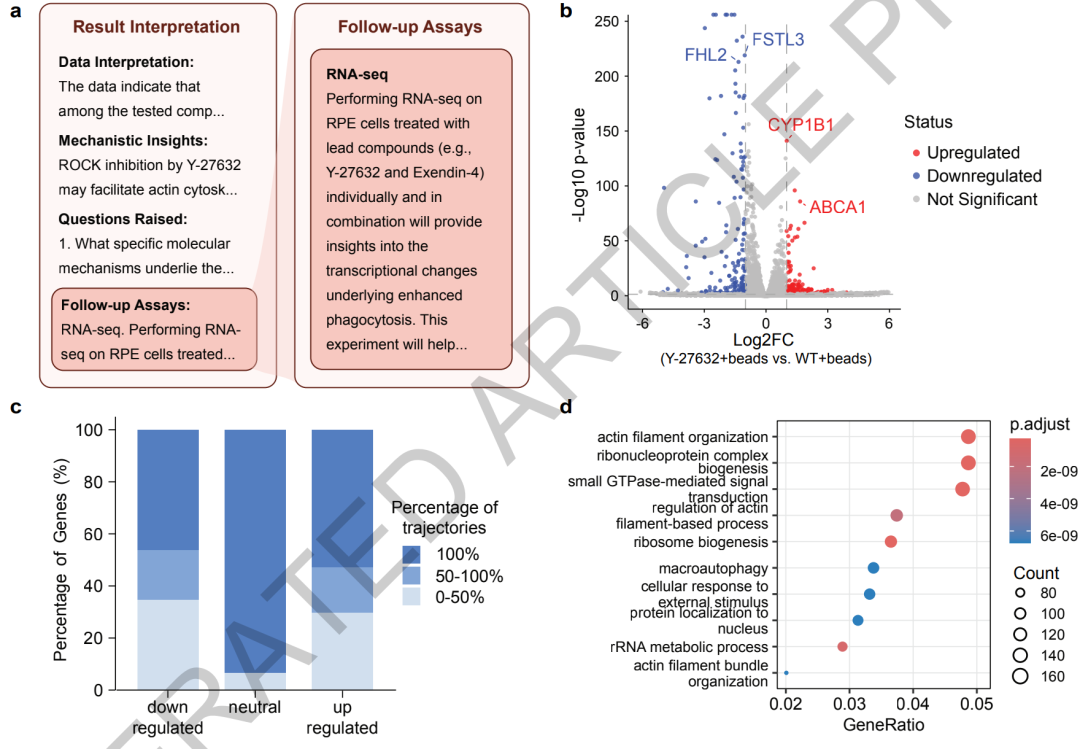
Robin entdeckte zudem, dass der circadiane Rhythmusregulator KL001 das Potenzial besitzt, die Phagozytosefunktion zu steigern, was einen neuen Forschungsansatz für die Behandlung der altersbedingten Makuladegeneration (AMD) eröffnet. Die anschließende Validierung des Transkriptoms bestätigte, dass Ripasudil die ABCA1-Expression stabil hochregulieren kann und klärte somit seinen zentralen Wirkmechanismus auf.
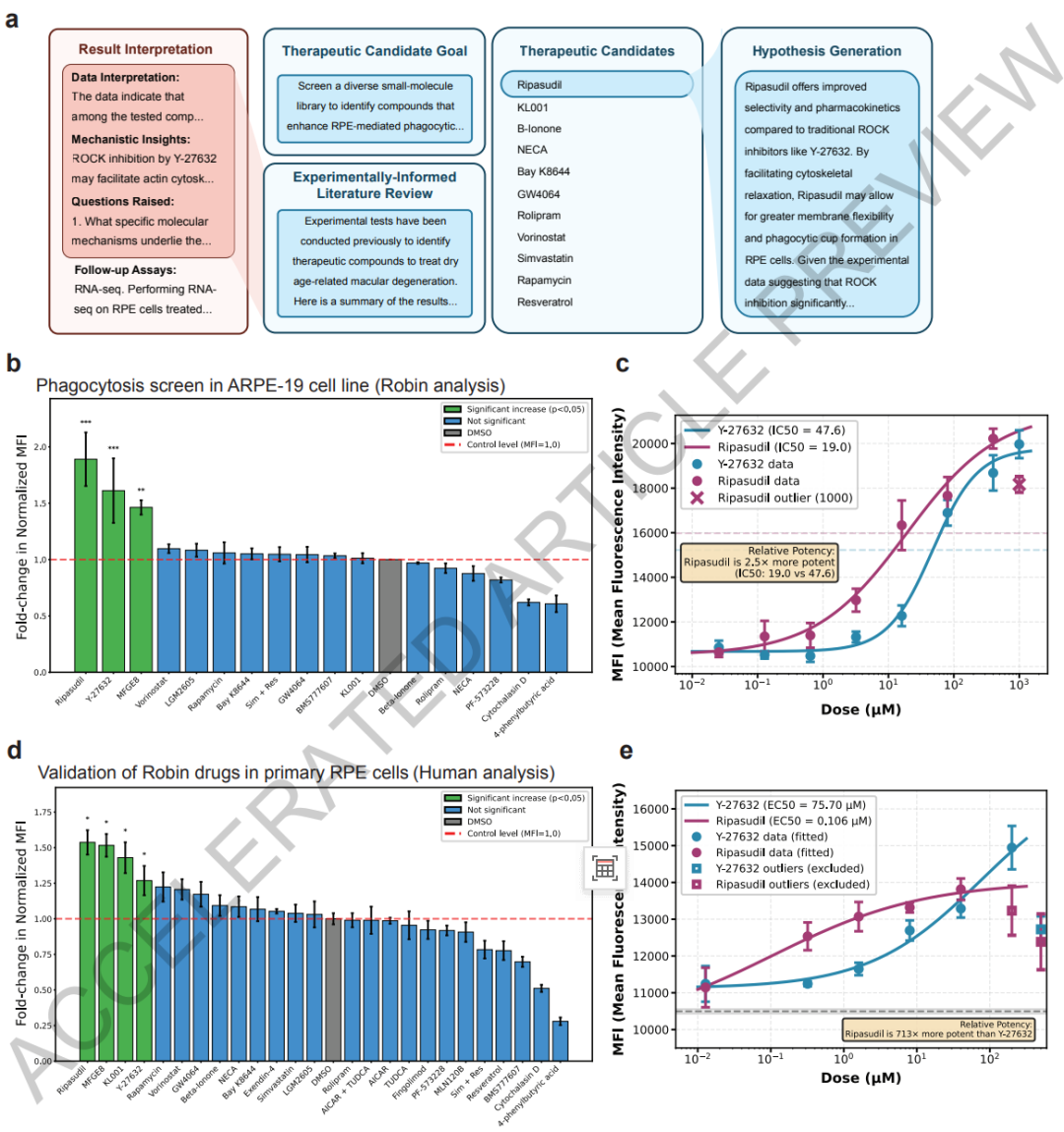
Im Vergleich zu allgemeinen KI-Forschungssystemen nutzte das Forschungsteam dieselben Anweisungen, um den OpenAI Deep Research Agent aufzurufen. Keiner der 17 generierten Wirkstoffkandidaten zeigte eine Steigerung der Phagozytoseaktivität, noch konnte der Kernmechanismus der ROCK-Hemmung aufgeklärt werden. Dies unterstreicht die Vorteile von Robin hinsichtlich seiner Anpassungsfähigkeit in spezifischen biomedizinischen Anwendungsbereichen.
Des Weiteren im BixBench-Benchmark-TestDie Gesamtgenauigkeit des Finch-Agenten erreichte 22,8±1,7%, was deutlich höher ist als die 1,6±1,2% des reinen großen Sprachmodells.Die Genauigkeitsraten für biostatistische Aufgaben erreichten 47,9 ± 1,51 TP3T, für die grundlegende Durchflusszytometrie-Analyse 1001 TP3T und für die RNA-Seq-Analyse 861 TP3T. Diese Ergebnisse zeigen, dass das speziell entwickelte Framework für Forschungsagenten die praktischen Fähigkeiten allgemeiner, großer Modelle in der Analyse biologischer Daten deutlich verbessern kann. Dennoch besteht bei komplexen, mehrstufigen Bioinformatik-Aufgaben noch Optimierungspotenzial.
Robin bietet zudem einen klaren Vorteil hinsichtlich Effizienz und Kosten. Forschungsstatistiken zeigen, dass die durchschnittlichen Kosten eines einzelnen, vollständigen Forschungsworkflows mit dem System nur etwa 10,76 US-Dollar betragen;Robin konnte die integrierte Analyse von 551 Dokumenten innerhalb von 30 Minuten abschließen.Die gleiche Arbeitsmenge würde manuell üblicherweise mehr als 800 Stunden in Anspruch nehmen. Insgesamt schließt das System einen einzelnen Recherchedurchlauf in weniger als 2 Stunden ab, was etwa 200-mal effizienter ist als herkömmliche manuelle Rechercheverfahren.
Letzte Worte
Robins Bedeutung reicht weit über die Entdeckung mehrerer potenzieller Wirkstoffkandidaten hinaus. Vor allem zeigt sie erstmals das Potenzial künstlicher Intelligenz auf, sich in den Lebenswissenschaften von einem „assistierenden Werkzeug“ zu einem „semi-autonomen Forschungssystem“ zu entwickeln. Natürlich sind solche Systeme noch weit davon entfernt, wirklich „autonome Wissenschaftler“ zu sein. Herausforderungen wie komplexe Versuchsplanung, das Verständnis biologischer Mechanismen über verschiedene Skalen hinweg und die Interpretierbarkeit von Ergebnissen hängen weiterhin maßgeblich von der Beteiligung von Fachexperten ab. Robins Entwicklung zeigt jedoch zumindest, dass KI nicht länger nur ein Werkzeug zur Effizienzsteigerung von Forschern ist, sondern zunehmend die Fähigkeit erlangt, selbst aktiv an wissenschaftlichen Entdeckungen mitzuwirken.








