Command Palette
Search for a command to run...
Anhand Von 220 Marinen Bakterienarten Rekonstruierten Wissenschaftler Mithilfe Eines Genomweiten Modells Das Heterotrophe Mikrobielle Klassifizierungssystem Und Identifizierten Dabei Acht Typen Von Stoffwechselflora.

Wälder gelten als die Lunge der Erde, der Ozean als ihr Herz. Die Weiten der Ozeane beherbergen Zehntausende von Mikroorganismen, die komplexe Gemeinschaften bilden. Durch ihre einzigartigen Stoffwechselspezialisierungen regulieren sie die Umwandlung organischer Substanz und steuern die Kohlenstofffixierung und -freisetzung. So beeinflussen sie maßgeblich die globalen Kohlenstoffkreisläufe, den Klimawandel und die marine Biodiversität. Heterotrophe Mikroorganismen im Meer fungieren dabei als „Reinigungseinheiten“ des marinen Ökosystems. Sie übernehmen die zentrale Aufgabe des Abbaus organischer Substanz und erhalten so die globalen Stoffkreisläufe und das ökologische Gleichgewicht aufrecht.
Lange Zeit wurden marine heterotrophe Mikroorganismen klassisch in zwei Hauptkategorien eingeteilt: kopiotrophe und oligotrophe. Erstere gedeihen in Umgebungen mit hohem Gehalt an organischer Substanz, während letztere in nährstoffarmen Umgebungen nur langsam überleben. Diese traditionelle „Dichotomie“, die viele Jahre lang Anwendung fand, hat der biogeochemischen Forschung zwar in gewissem Maße geholfen, weist aber auch erhebliche Schwächen auf: Die Wachstumsrate lässt sich nicht mit Substratnutzungspräferenzen und metabolischen Nischen gleichsetzen. So wie man menschliche Ernährungsgewohnheiten nicht allein anhand der verzehrten Nahrungsmenge klassifizieren kann, ist die individuelle Ernährungspräferenz der entscheidende Faktor für die Geschwindigkeit des Abbaus organischer Substanz und damit für die Richtung des Kohlenstoffkreislaufs.
Um diesem Problem zu begegnen, analysierte ein Team der University of Southern California mithilfe der Ocean Microbial Database (OMD) riesige Mengen mariner Bakteriengenome und nutzte dafür genomweite Stoffwechselmodelle (GEMs). Durch die Quantifizierung der Empfindlichkeit von Mikroorganismen gegenüber der Nutzung von elf verschiedenen organischen Substraten gelang es ihnen schließlich, das traditionelle „Dichotomie“-Modell zu überwinden.Es wurden acht Kategorien differenzierter metabolischer Mikrobiota identifiziert: eine Kategorie schnellwachsender eutropher Mikrobiota, drei Kategorien substratspezifischer langsamwachsender oligotropher Mikrobiota und vier Kategorien substratspezialisierter intermediär wachsender Mikrobiota.
Die Ergebnisse mit dem Titel „Definition metabolischer Nischen für marine mikrobielle Heterotrophe“ wurden in Science Advances veröffentlicht.
Forschungshighlights:
* Es löst sich vom klassischen "Dichotomie"-Ansatz und verankert mikrobiell-spezifische metabolische Nischen auf der Grundlage tatsächlicher Stoffwechselstrategien und Substratpräferenzen.
* Basierend auf acht funktionellen mikrobiellen Gemeinschaften deckt diese Studie systematisch die Wachstumsmuster, Ressourcenkonkurrenzmodelle und die globale geographische Verteilung mariner heterotropher Mikroorganismen auf und verdeutlicht so den intrinsischen Mechanismus, durch den Mikroorganismen den marinen Kohlenstoffkreislauf antreiben.
* Um die Forschungslücken hinsichtlich der Beteiligung mariner heterotropher Mikroorganismen am globalen Kohlenstoffkreislauf zu schließen und um verfeinerte Verbesserungsideen und Parameterschemata für biogeochemische Modelle bereitzustellen.

Papieradresse:
https://www.science.org/doi/10.1126/sciadv.adz0537
Datensatz: Umfasst 220 verschiedene Kategorien von Meeresbakterien
Diese Studie basiert auf einem umfangreichen Datensatz mariner mikrobieller Genome, der aus der OMD-Datenbank stammt, die auf der Plattform microbiomics.io gehostet wird.Die Datenbank enthält ungefähr 35.000 mikrobielle Genome.Dazu gehören metagenomisch zusammengesetzte Genome, durch Einzelzellamplifikation entstandene Genome sowie Genome künstlich isolierter und kultivierter Stämme.
Diese Studie umfasste ausschließlich bakterielle Genome mit einer Integrität von > 801 TP3T und einer Kontaminationsrate von < 51 TP3T. Die beiden Wertesätze wurden anhand der Mittelwerte der Software CheckM bzw. Anvi'o berechnet. Anschließend nutzten die Forscher Metadaten aus der OMD-Datenbank und die Software dRep, um Redundanzen in den Genomen mithilfe eines Schwellenwerts für die durchschnittliche Nukleotididentität (ANI) von 951 TP3T zu entfernen.
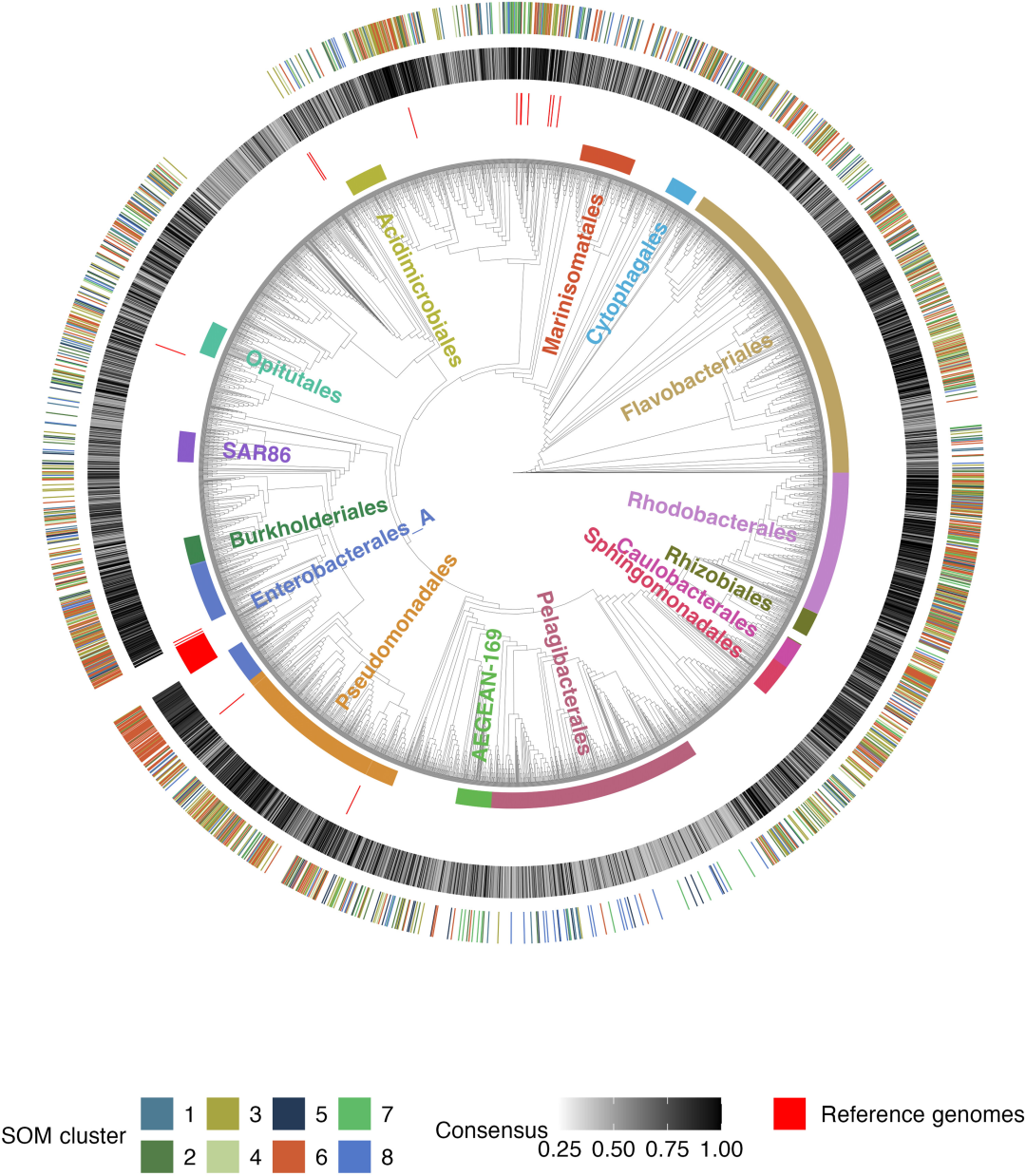
Nach dem Entfernen von 180 photosynthetischen autotrophen Cyanobakterien (Exogruppen),Nach Entfernung redundanter Sequenzen wurden schließlich 3.738 qualitativ hochwertige heterotrophe Bakteriengenome gewonnen.Dies stellt den grundlegenden analytischen Datensatz für diese Studie dar. Der Datensatz umfasst 220 verschiedene Kategorien mariner Bakterien, von denen 14 Gruppen ≥ 50 Genome enthalten.
Beim Aufbau des phylogenetischen Baums wurden neben 180 Cyanobakterien-Genomen als Außengruppen 66 bakterielle Referenzgenome aus der BiGG-Datenbank hinzugefügt, wodurch sich die Gesamtzahl der Genome auf 3.984 erhöhte. Acht Genome wurden aufgrund unzureichender Übereinstimmung der Zielgene (Einzelkopien) aus dem phylogenetischen Baum entfernt.Letztendlich wurden 3.976 Pflanzen zur Konstruktion von Entwicklungsbäumen verwendet.Die vollständigen Genomklassifizierungsinformationen wurden mithilfe der GTDB-Tk v2.1.0- und GTDB r214-Datenbanken auf dem Baum gekennzeichnet.
Selbstorganisierende neuronale Netze werden zur Klassifizierung metabolischer Nischen verwendet.
Um die Grenzen des traditionellen „Dichotomie“-Ansatzes zu überwinden,Diese Studie integriert Genomik, eingeschränkte Stoffwechselsimulation und unüberwachte maschinelle Lernverfahren, um einen vollständigen analytischen Rahmen von genetischen Informationen bis hin zur mikrobiellen Ökotypisierung zu schaffen.Die metabolische Modellierung, die Quantifizierung der Substratsensitivität und die Clusterung der mikrobiellen Gemeinschaft wurden hierarchisch unter Verwendung verschiedener Arten von Feldmessungen und globaler Umweltdatensätze durchgeführt.
Modellierung und Qualitätskontrolle
In der Phase der Modellbildung wandten die Forscher eine integrierte Modellierungsstrategie an.Mit der Software CarveMe v1.5.1 wurden 60 unabhängige Stoffwechselmodelle (Modellsätze) für jeden der 3738 marinen heterotrophen Bakterienstämme erstellt.
Das Modellierungsprinzip der CarveMe-Software basiert auf einer allgemeinen Architektur metabolischer Modelle. Anhand des Vorhandenseins oder Fehlens jeder biochemischen Reaktion in den eingegebenen Genomannotationsinformationen werden jedem Reaktionsschritt Gewichte zugewiesen. Dadurch wird das allgemeine Modell initialisiert und das entsprechende metabolische Modell des Genoms vorhergesagt. In dieser Studie wurde die Anzahl der erforderlichen Modellwiederholungen zur Abdeckung des Reaktionsprofils eines einzelnen Genoms umfassend untersucht. Die Ergebnisse zeigen, dass die Gesamtzahl der neu hinzugefügten Reaktionen relativ konstant bleibt, sobald die Anzahl der Modelle im Ensemble etwa 60 erreicht.Dies beweist, dass ein Satz von 60 Modellen die überwiegende Mehrheit der möglichen Stoffwechselmodellkombinationen für ein einzelnes Genom abdecken kann.
Um die Qualität des von CarveMe generierten Stoffwechselmodells zu quantifizieren, wurde in der Studie erstmals ein Konsistenz-Score-Index C als Bewertungsmetrik entwickelt, wie in der folgenden Formel dargestellt:
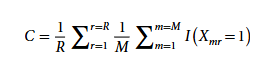
In der Formel repräsentiert Xmr die „Präsenz-Deletions-Matrix“ der Ensemble-Modellantwort in den M unabhängig erstellten Modellen; r bezeichnet eine einzelne biochemische Antwort; R ist die Gesamtzahl der Antworten im gesamten Modellset; und I ist eine Indikatorfunktion, die angibt, ob die Antwort r im m-ten Ensemble-Teilmodell vorhanden ist. In der nachfolgenden Analyse wurden nur Genomproben mit einem Konsistenzwert ≥ 0,8 berücksichtigt, insgesamt 1.578 Genome.
Bewertung der Stoffwechselstrategie
Forscher definieren Stoffwechselstrategien als die bevorzugten Substrate für das Wachstum eines Organismus, und ihre Methodik beinhaltet die Interpretation dieser Strategien durch eine Reihe von Sensitivitätsanalysen.
Speziell,Die Forscher verwendeten das Flux Balance Analysis (FBA) Toolkit im Softwarepaket COBRApy v0.25.0, um Wachstumssensitivitätstests am CarveMe-Modell sowohl unter „substratausreichenden“ als auch unter „substratbegrenzten“ Substratversorgungsbedingungen durchzuführen. Der Zustand der „Substratlimitierung“ wird erreicht, indem der verfügbare Fluss einer bestimmten Verbindungsklasse auf 50% der Menge reduziert wird, die der Organismus unter „Substratausreichenden“ Bedingungen aufnehmen würde.
Um die Unterschiede in den Substratanforderungen zwischen verschiedenen Modellen zu quantifizieren, wird ein Sensitivitätskoeffizientenindex S vorgeschlagen, wie unten dargestellt:
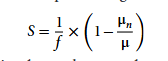
In der Formel steht μn für die vorhergesagte Wachstumsrate unter Substratlimitierung für Typ n, μ für die vorhergesagte Wachstumsrate unter Substratüberschuss und f für den Substratlimitierungskoeffizienten (in dieser Studie mit 0,5 angenommen). Der Sensitivitätskoeffizient S liegt im Bereich von 0 bis 1. Das bedeutet: Sinkt die vom Modell berechnete Wachstumsrate um 50%, nachdem das Substratangebot um 50% reduziert wurde, so ist dieser Substrattyp ein vollständig limitierender Faktor für das Wachstum des Organismus (Wachstumssensitivität = 1). Bleibt die modellierte Wachstumsrate hingegen unverändert, so wird das Wachstum des Organismus durch das Angebot dieses Substrattyps nicht beeinflusst (Wachstumssensitivität = 0).
Wenn das Verhältnis des Ausmaßes der Substratbeschränkung zum Ausmaß der Abnahme der Wachstumsrate ≥ 0,8 beträgt (S ≥ 0,8), wird davon ausgegangen, dass das Modell eine signifikante Wachstumssensitivität gegenüber solchen Substraten aufweist.
Clusteranalyse im unüberwachten maschinellen Lernen
Im maschinellen Lernteil der Studie wurden selbstorganisierende Karten (SOMs) verwendet, um metabolische Nischen abzugrenzen. SOMs ist ein unüberwachter Algorithmus des maschinellen Lernens, der die Dimensionalität massiver hochdimensionaler Datensätze auf einen zweidimensionalen Gitterraum mit topologischer Struktur reduzieren kann.
Vor der Clusteranalyse führten die Forscher eine weitere Datenprüfung der 1.578 zuvor ermittelten Genome durch und zählten die Varianz der Wachstumssensitivität verschiedener Metaboliten in allen 60 Submodellen. Sie entfernten 100 Genome mit einer Gesamtvarianz der Substratsensitivität von > 0,1, sodass 1.478 Genome verblieben. Insgesamt wurden 88.680 gültige Datensätze (1.478 Genome x 60 Ensemble-Modelle) für die SOM-Clusteranalyse verwendet. Jeder Datenpunkt enthielt 11 Indikatoren für die charakteristische metabolische Sensitivität.
Für die Verarbeitung der standardisierten Daten zur Vorhersage des zusammengesetzten Flusses wurde in dieser Studie die Software Kohonen v3.0.12 verwendet. Es wurden 1500 Iterationen auf einem 20 × 20 toroidalen hexagonalen Gitter durchgeführt (wobei die räumliche Distanz anhand der euklidischen Standarddistanz charakterisiert wurde). Die Lernrate wurde auf (0,025, 0,01) eingestellt, und der Nachbarschaftsradius wurde aus dem Standardwert der Software übernommen.
Nach ausreichendem Training und basierend auf der Konsistenz der Vorhersageergebnisse zur Wachstumshemmung wird der K-Means-Clustering-Algorithmus angewendet.Die SOM-Karte wurde schließlich in 8 differentielle Cluster unterteilt.
Nach der Clusterung wurde zur Beurteilung der Unterschiede in der maximalen Wachstumsrate der dCUB-Wert aller 1478 Genome mithilfe von gRodon berechnet, um schnell- und langsamwachsende Bakterien zu klassifizieren. Basierend auf 1209 Metagenomen aus verschiedenen Datensätzen, darunter Tara Oceans, BioGeoTraces und Malaspina sowie einem globalen ASV-Datensatz von McNichol et al., wurde die globale geografische Verteilung der acht Bakteriengemeinschaften validiert.
Acht Stoffwechselcluster wurden anhand ihrer Substratpräferenz und Wachstumsrate klassifiziert.
Die Studie präsentiert diverse experimentelle Ergebnisse, die nicht nur die Leistungsfähigkeit des Modells bestätigen, sondern vor allem den traditionellen Ansatz der „Dichotomie“ durchbrechen, eine völlig neue Klassifizierungslogik vorschlagen und die intrinsische Beziehung zwischen Substratpräferenz und metabolischer Nische herstellen.
Ergebnisse der Modellvalidierung
Forscher validierten die Genauigkeit des CarveMe-Modells hinsichtlich der Substratpräferenzen in groß angelegten Kulturexperimenten zu den Kohlenstoffquellenpräferenzen von 186 marinen Mikroorganismen. Konkret erstellten sie ein CarveMe-Modell für die von Gralka et al. untersuchten Genome und führten entsprechende In-silico-Experimente mit FBA durch, um das Wachstum dieser Mikroorganismen unter denselben Kohlenstoffquellenbedingungen zu testen.
Die Ergebnisse zeigen, dassIm Vergleich zu den experimentellen Daten in der Literatur erreichten die Modellvorhersageergebnisse eine Genauigkeit von 75,51 TP3T und eine Genauigkeit von 87,41 TP3T.Um zu beurteilen, ob dieses Ergebnis wesentlich besser war als eine zufällige Vorhersage, testeten die Forscher es, indem sie eine Bootstrap-Analyse der zufälligen Vorhersage durchführten. Die Ergebnisse zeigten, dass die Genauigkeit des Modells wesentlich höher war als die des Zufallsniveaus.
Ergebnisse der Typisierung von 8 Bakterienarten
Basierend auf 1.478 Genomsequenzen und 11 sensitiven Indikatoren,Die Studie identifizierte acht unterschiedlich exprimierte metabolische mikrobielle Gemeinschaften mittels SOM-Clustering und kategorisierte sie anhand ihrer Wachstumsraten in drei Hauptgruppen – schnell, mittel und langsam.Im Einzelnen (wie in der Abbildung unten dargestellt):
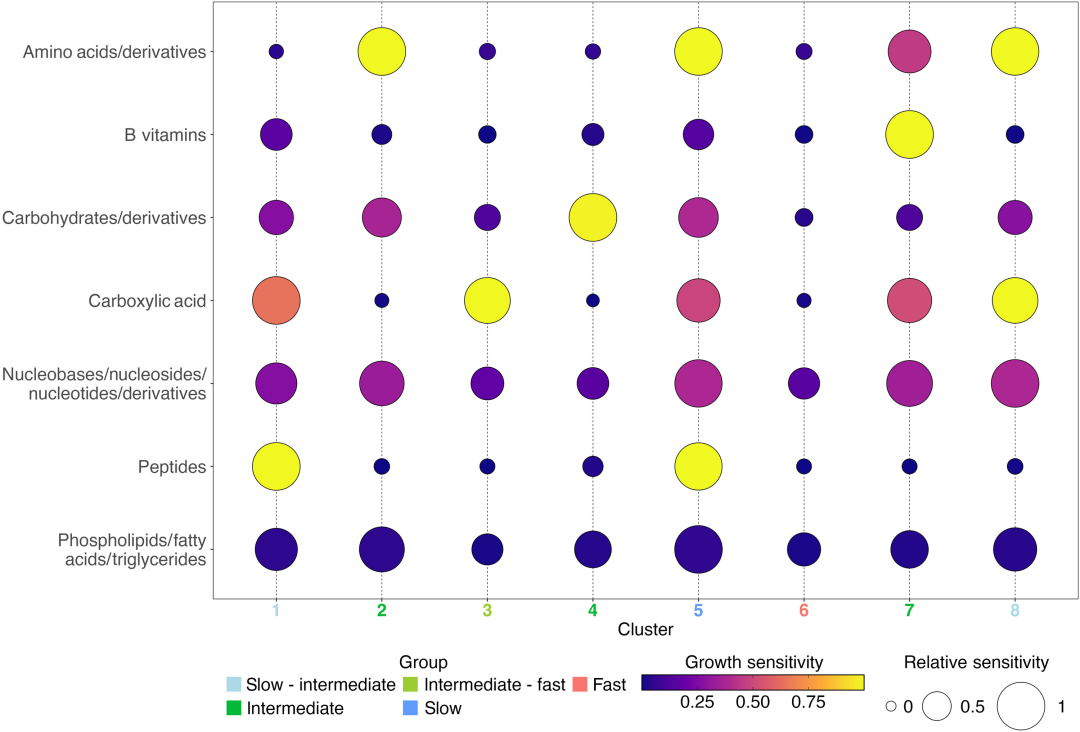
Vergleich der durchschnittlichen Wachstumsempfindlichkeit von 8 SOM-Clustern
Kategorie 1: Schnellwachsende eutrophe Mikrobiota (Cluster 6): Dies ist eine typische eutrophe Mikrobiota, deren 79.5%-Genom eine maximale Wachstumsrate vorhersagt, die über der Schwelle für langsames Wachstum liegt (dCUB < -0,08 ist die Schwelle für schnelles Wachstum; ein kleinerer dCUB-Wert deutet auf schnelleres Wachstum hin). Taxonomisch gesehen gehören Enterobacterales, Flavobacteriales, Rhodobacterales und Pseudomonadale zu den typischen Vertretern von Cluster 6. Diese Mikrobiota wird am wenigsten von Substraten beeinflusst; das Fehlen einer der 11 getesteten Verbindungen hemmte ihr Wachstum nicht.
Drei substratspezifische, langsam wachsende, oligotrophe Bakteriengruppen (Cluster 1, Cluster 5 und Cluster 8): dCUB = -0,111 für diese Gruppen. Cluster 5 (61,81 TP3T) weist die niedrigste maximale Wachstumsrate auf; typische Vertreter sind Opitutales (Verrucomicrobiota) und Pelagibacterales, deren Anreicherung in dieser Gruppe 4351 TP3T bzw. 3621 TP3T erreicht.
Vier substratspezifische Bakteriengruppen mit intermediärem Wachstum (Cluster 2, 3, 4 und 7): Diese Gruppen wiesen erwartungsgemäß signifikant niedrigere Wachstumsraten als Cluster 6, aber signifikant höhere als Cluster 5 auf. Cluster 3 zeigte eine signifikant höhere Wachstumsrate als die Cluster 1 und 8. Jede dieser vier Bakteriengruppen mit intermediärem Wachstum reagierte empfindlich auf nur eine Verbindungsklasse: Aminosäuren, Carbonsäuren, Kohlenhydrate und B-Vitamine.
Die Eigenschaften von mikrobiellen Gemeinschaften mit mittlerem Wachstumstempo bestätigen zudem eine aktuelle Studie, die nahelegt, dass die dominanten heterotrophen Mikroorganismengruppen im marinen Untergrund langsam wachsende, eutrophe Bakterien sein könnten. Diese Erkenntnis kann als Grundlage für die Klassifizierung dieser Mikroorganismen in metabolische Funktionsgruppen dienen, beispielsweise für die Präferenz von Cluster 4 für Kohlenhydrate.
Letzte Worte
Zusammenfassend lässt sich sagen, dass diese Studie mit dem jahrzehntealten Dichotomie-Modell von eutrophen/oligotrophen Nischen bricht und ein achtstufiges Klassifizierungssystem für metabolische Nischen etabliert, das auf dem Wesen der Gen- und Substratnutzung basiert. Dadurch wird die inhärente Bindung zwischen den fünf Kategorien und den physiologischen Funktionen aufgehoben.
Darüber hinaus vereinfacht dieses neue Klassifizierungssystem die komplexe Struktur der Vielzahl heterotropher Mikroorganismen im Ozean. Zukünftig kann es in globale biogeochemische Modelle integriert werden, wodurch die Notwendigkeit entfällt, Zehntausende mariner Bakterien einzeln zu erfassen. Es ermöglicht die Ableitung des Abbaus mariner organischer Substanz und von Veränderungen im Kohlenstoffhaushalt anhand von nur acht funktionellen Parametern und bietet somit ein völlig neues theoretisches Werkzeug zur Bewertung der Entwicklung des ozeanischen Kohlenstoffkreislaufs vor dem Hintergrund der globalen Erwärmung.








