Command Palette
Search for a command to run...
Ein Deutsches Team Hat Einen Neuen Durchbruch in Der Biomedizinischen Forschung Mit Kleinen Stichproben Erzielt, Indem Es Generative KI-Modelle Zur Datenerweiterung einsetzte. Dadurch Könnte Die Anzahl Der Benötigten Labortiere Pro TP3T Um 30 Bis 50 Reduziert werden.

Die in Tierversuchen nachgewiesenen „wirksamen therapeutischen Effekte“ lassen sich in klinischen Studien häufig nicht reproduzieren, wobei eine unzureichende Stichprobengröße einer der Hauptgründe ist. Zahlreiche weitere Einschränkungen, darunter ethische Richtlinien, Versuchskosten und Forschungsbedingungen, verschärfen das Problem zusätzlich.Die präklinische biomedizinische Forschung steht oft vor der Herausforderung, groß angelegte Tierversuche durchzuführen, was direkt zu einer unzureichenden statistischen Aussagekraft führt.Forscher sind nicht in der Lage, zuverlässige biologische Signale zu extrahieren und sind stark anfällig für falsch positive Ergebnisse, was die Übertragung der Grundlagenforschung in klinische Anwendungen ernsthaft behindert.
Um dieser Herausforderung zu begegnen, haben Wissenschaftler versucht, Forschungsdaten mithilfe von Methoden wie Metaanalyse und Datenzusammenführung zu integrieren.Allerdings sind diese Methoden stark von der Vergleichbarkeit der Versuchsaufbauten, der Nachweisindikatoren und der Arbeitsabläufe zwischen den verschiedenen Studien abhängig.Sein praktischer Anwendungsbereich ist äußerst begrenzt.
In den letzten Jahren hat die generative künstliche Intelligenz einen neuartigen Ansatz für die Forschung mit kleinen Stichproben geboten: Durch das Erlernen der inhärenten Verteilungsstruktur der Originaldaten generiert sie synthetische Daten, um den Stichprobenumfang zu erweitern. Allerdings weisen allgemeine generative Modelle erhebliche Schwächen auf:Wenn die Originaldaten zufällige Fehler enthalten, verstärkt das Modell das Rauschen zusätzlich und erzeugt eine große Anzahl falsch positiver Ergebnisse, was die Glaubwürdigkeit der Forschungsergebnisse mindert.Die Unterdrückung der Fehlerfortpflanzung bei der Datengenerierung hat sich zu einem zentralen Engpass für die Anwendung generativer KI im biomedizinischen Bereich entwickelt.
Um dieses kritische Problem anzugehen, entwickelte ein gemeinsames Forschungsteam der Universität Frankfurt und des Fraunhofer-Instituts für ITMP genESOM –Generative KI-Modelle, die auf emergenten selbstorganisierenden Karten basieren, sind speziell für biomedizinische Daten mit kleinen Stichproben konzipiert.Die Kerninnovation dieses Modells liegt in der Entkopplung des Strukturlernens vom Datengenerierungsprozess. Es verhindert die Fehlerfortpflanzung durch Dimensionsanpassung und führt eine negative Kontrollvariable ein, um die Qualität der Datengenerierung in Echtzeit zu überwachen. Das Forschungsteam nutzte präklinische Lipidomdaten von Patienten mit Multipler Sklerose als Untersuchungsobjekt, reduzierte zunächst die Stichprobengröße künstlich auf die statistische Ausfallschwelle und führte anschließend mit genESOM eine Datenaugmentation durch.
Die Ergebnisse bestätigen, dass diese Methode wichtige biologische Signale, die in kleinen Stichproben verloren gehen, effektiv wiederherstellen und gleichzeitig falsch-positive Ergebnisse streng kontrollieren kann. Sie bietet somit einen zuverlässigen neuen Ansatz für die biomedizinische Forschung mit kleinen Stichproben. Darüber hinaus wird erwartet, dass dieses Modell in explorativen Forschungsszenarien die Anzahl der benötigten Versuchstiere um etwa 301–501 Tiere reduziert, ohne die Reproduzierbarkeit und wissenschaftliche Validität der Ergebnisse zu beeinträchtigen.
Die zugehörigen Forschungsergebnisse mit dem Titel „Selbstorganisierende neuronale Netzwerk-basierte generative KI mit eingebetteter Fehlerinflationskontrolle verbessert die effektive Wissensextraktion aus präklinischen Studien mit reduzierter Stichprobengröße“ wurden in Pharmacological Research veröffentlicht.
Forschungshighlights:
* Der integrierte datengesteuerte Fehlerkontrollmechanismus unterdrückt effektiv die Inflation falsch positiver Ergebnisse, im Gegensatz zu unbeschränkten Methoden wie GAN.
* Wichtige Lipidsignale (wie z. B. Lysophosphatidsäure) konnten nach der Reduzierung der Probenmenge erfolgreich wiederhergestellt werden, ohne dass die Rate falsch positiver Ergebnisse anstieg.
* Es kann den Tierverbrauch um 30–50 % reduzieren und dient als ergänzendes Analyseinstrument, wobei sowohl die Robustheit der Forschung als auch die ethischen 3R-Prinzipien berücksichtigt werden.

Lesen Sie das Dokument:
https://www.sciencedirect.com/science/article/pii/S1043661826000745
Datensätze: Von vollständigen Experimenten bis hin zu statistischen Fehlern bei kleinen Stichproben
Die Daten für diese Studie stammen aus einer öffentlich publizierten präklinischen Tierstudie zu Multipler Sklerose.In der Studie wurden SJL/J-Mäuse verwendet, um ein rezidivierend-remittierendes experimentelles Autoimmunenzephalomyelitis-Modell (EAE) zu etablieren.Ziel der Studie ist es, die Mechanismen der Neuroinflammation aufzuklären und die therapeutische Wirksamkeit des zugelassenen Medikaments Fingolimod zu bestätigen.
Hinweis: Fingolimod ist ein Modulator des Sphingosin-1-Phosphat-Rezeptors, der durch die Regulierung des Sphingolipidstoffwechsels in Immunsignalwege eingreifen kann. Es ist ein häufig eingesetztes Medikament in der klinischen Behandlung von Multipler Sklerose.
Das Experiment umfasste 26 acht Wochen alte weibliche Mäuse, die zufällig in drei Gruppen eingeteilt wurden: eine Kontrollgruppe (ohne Behandlung), eine EAE-Modellgruppe und eine EAE-Gruppe mit Fingolimod-Behandlung. Die Behandlungsgruppe erhielt das Medikament ab dem 18. Tag nach der Immunisierung über das Trinkwasser in einer Dosis von 0,5 mg/kg/Tag.
Das Forschungsteam sammelte gleichzeitig Daten auf Verhaltens- und molekularer Ebene:Verhaltensindikatoren umfassen motorische Fähigkeiten, körperliche Koordination und Sozialverhalten; auf molekularer Ebene wurde die LC-MS/MS-basierte gezielte Quantifizierungstechnologie eingesetzt, um die Konzentration von 62 Lipidmediatoren in vier Geweben zu bestimmen: Plasma, Kleinhirn, Hippocampus und präfrontaler Cortex. Diese decken vier Hauptkategorien ab: Lysophosphatidsäure, Ceramide, Sphingolipide und Endocannabinoide.Abschließend wurde eine Standarddatenmatrix der „individuellen Maus × Lipidcharakteristika“ erstellt.
Vor der DatenanalyseDas Forschungsteam führte eine logarithmische Transformation der Lipidkonzentrationsdaten durch, um diese an die Verteilungsannahmen der statistischen Analyse anzupassen.Die fehlenden Werte in den Originaldaten (5.3%) wurden nach der multimodalen Datenausrichtung mithilfe des Random-Forest-Algorithmus (missForest) ergänzt. Anschließend wurde eine einfaktorielle Varianzanalyse (ANOVA) mit 62 Lipidindikatoren durchgeführt und die Šidák-Korrektur zur Kontrolle multipler Testfehler angewendet. Parallel dazu wurden drei Modelle des maschinellen Lernens – Random Forest, Support Vector Machine und k-Nearest Neighbors – zur Kreuzvalidierung der Stabilität der biologischen Signale in den Daten hinsichtlich zweier Dimensionen eingesetzt: der Signifikanz von Unterschieden zwischen Gruppen und der Klassifizierungsvorhersagefähigkeit.
Nach Abschluss der Basisanalyse wurde ein zentrales Validierungsexperiment durchgeführt: Die Stichprobengröße wurde systematisch reduziert, um den kritischen Wert für statistisches Versagen bei kleinen Stichproben zu ermitteln. Die Forscher reduzierten schrittweise die Anzahl der Mäuse in jeder Gruppe und wiederholten nach jeder Reduzierung das gesamte Analyseverfahren. Die Ergebnisse zeigten, dass bei einer Reduzierung der Stichprobengröße auf 6 Mäuse pro GruppeDas vollständige Verschwinden aller signifikanten statistischen Ergebnisse aus den Originaldaten dient als Maßstab für die Bewertung der Datenerweiterungsfähigkeiten von genESOM.—In Szenarien mit kleinen Stichproben, in denen statistische Methoden völlig wirkungslos sind, soll überprüft werden, ob KI biologische Signale wiederherstellen kann, die durch Rauschen überdeckt wurden.
Generative KI, speziell entwickelt für biomedizinische Daten mit kleinen Stichproben
Herkömmliche generative Modelle stehen bei der Verarbeitung kleiner Stichprobendaten vor einem Dilemma: Entweder sind die generierten Daten informationsarm und können die ursprünglichen biologischen Signale nicht rekonstruieren, oder sie sind mit Rauschen überladen, was zu einer hohen Anzahl falsch positiver Ergebnisse führt. Das Kernkonzept von genESOM besteht darin, einen präzisen Ausgleichsmechanismus zwischen diesen beiden Faktoren zu etablieren und so eine sichere und interpretierbare Erweiterung kleiner Stichprobendaten zu ermöglichen.
genESOM basiert auf dem neuronalen Netzwerk Emergent Self-Organizing Map (ESOM) und erzielt im Vergleich zur klassischen Self-Organizing Map (SOM) zwei wesentliche Verbesserungen:Erstens,Die Neuronen sind in einem zweidimensionalen kreisförmigen Gitter angeordnet, um die Nachbarschaftsstrukturbeziehungen der hochdimensionalen Daten bestmöglich zu erhalten.Zweitens,Die Hinzunahme einer dritten Dimension, die den Untergruppenabstand und den Projektionsfehler kodiert, verbessert die Genauigkeit der Identifizierung potenzieller Clusterstrukturen signifikant.
Nach Standardisierung und Entfernung fehlender Werte werden die Daten zur Trainingsplanung auf das ESOM-Netzwerk projiziert. Das Modell wählt kontinuierlich für jede Stichprobe das optimale Neuron aus, passt die Neuronengewichte dynamisch an und reduziert die Lernrate schrittweise, um die Stabilität des Trainings zu gewährleisten. Nach dem Training gibt das Modell zwei Kernmatrizen aus: Die U-Matrix charakterisiert den Abstand zwischen den Neuronen und identifiziert Clustergrenzen; die P-Matrix dient der statistischen Analyse der lokalen Datendichte und bildet die Grundlage für die Generierung synthetischer Daten. Der Radiusparameter, der den Bereich der synthetischen Datengenerierung steuert, wird automatisch durch Anpassen einer Distanzverteilung mithilfe eines Gaußschen Mischungsmodells bestimmt, ohne dass ein manueller Eingriff erforderlich ist.
Das bahnbrechendste Designmerkmal von genESOM ist die vollständige Trennung der Prozesse des Strukturlernens und der Datengenerierung.Das Modell lernt zunächst selbstständig die Repräsentation der inhärenten Datenstruktur und generiert anschließend synthetische Daten basierend auf dieser stabilen Struktur. Dadurch wird die Anhäufung von Fehlern aus den beiden Schritten vermieden. Wichtiger noch: Das Modell kann Permutationsvariablen als Negativkontrollen einführen, um in Echtzeit zu überwachen, ob die Bedeutung von Merkmalen übermäßig verstärkt wird. Sobald eine Fehleranhäufung festgestellt wird, wird die Datenerweiterung sofort und automatisch gestoppt. Dies minimiert das Risiko von Überanpassung und falsch positiven Ergebnissen.
In dieser Studie verwendete das Forschungsteam ein sicheres Anreicherungsverhältnis von 1:1 (eine synthetische Probe wurde aus jeder Originalprobe erzeugt), um die Stichprobengröße in jeder Gruppe von 6 auf 12 zu erhöhen. Nach der AnreicherungAnhand der Originaldaten wird ein vollständiger Satz statistischer und maschineller Lernanalysen durchgeführt, um den Effekt der Signalwiederherstellung quantitativ zu bewerten.In der Studie wurde genESOM direkt mit zwei gängigen generativen Methoden verglichen: dem Gaussian Mixture Model (GMM) und dem Conditional Table Generative Adversarial Network (CT-GAN). Als Kernindikatoren dienten die Falsch-Positiv-Rate, die Falsch-Negativ-Rate und die Wiederherstellungsrate des Originalsignals, um die Vorteile des Modells zu überprüfen.
Es ist herkömmlichen Generierungsmethoden in Szenarien mit kleinen Stichproben deutlich überlegen.
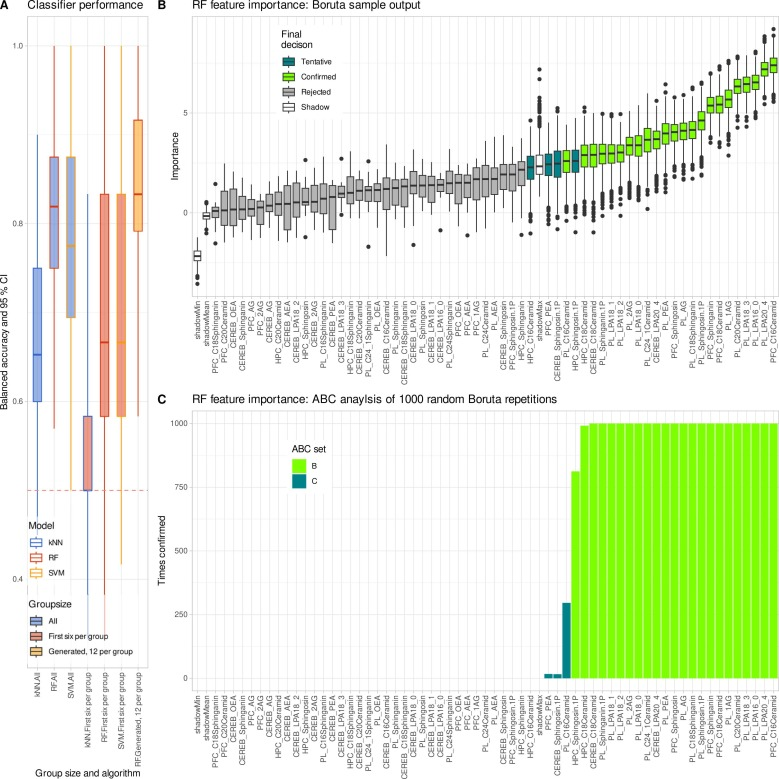
Wie die Abbildung unten zeigt, ergeben sich aus der Analyse des vollständigen Originaldatensatzes signifikante Unterschiede zwischen den Gruppen bei 27 der 62 Lipidvariablen, wobei die Veränderungen der Lysophosphatidylcholin-Lipide am deutlichsten ausgeprägt sind. Dieses Ergebnis deckt sich weitgehend mit früheren Forschungsergebnissen zu Multipler Sklerose. Gleichzeitig klassifiziert das Random-Forest-Modell die Proben mit einer Genauigkeit, die weit über der Zufallswahrscheinlichkeit liegt, was diese beiden Ergebnisse bestätigt.Dies bestätigt das Vorhandensein stabiler und zuverlässiger biologischer Signale in den Originaldaten.
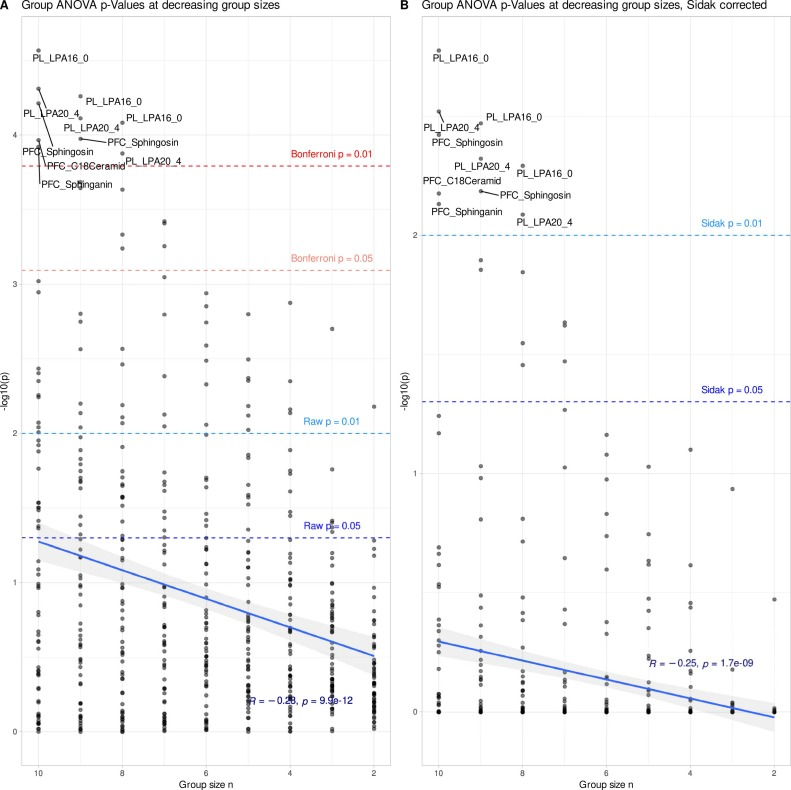
Als die Stichprobengröße in jeder Gruppe jedoch auf 6 Tiere reduziert wurde (siehe Abbildung unten), veränderten sich die Dateneigenschaften dramatisch: Nach Korrekturen mehrerer Validierungstests war die statistische Signifikanz aller Lipidindikatoren nicht mehr gegeben, und auch die Klassifizierungseffizienz des Random Forest sank deutlich. Es ist wichtig zu betonen, dass dies nicht bedeutet, dass die biologischen Effekte tatsächlich verschwunden sind.Stattdessen führt die geringe Stichprobengröße zu einer unzureichenden statistischen Aussagekraft, und das eigentliche Signal wird vom Rauschen überlagert.
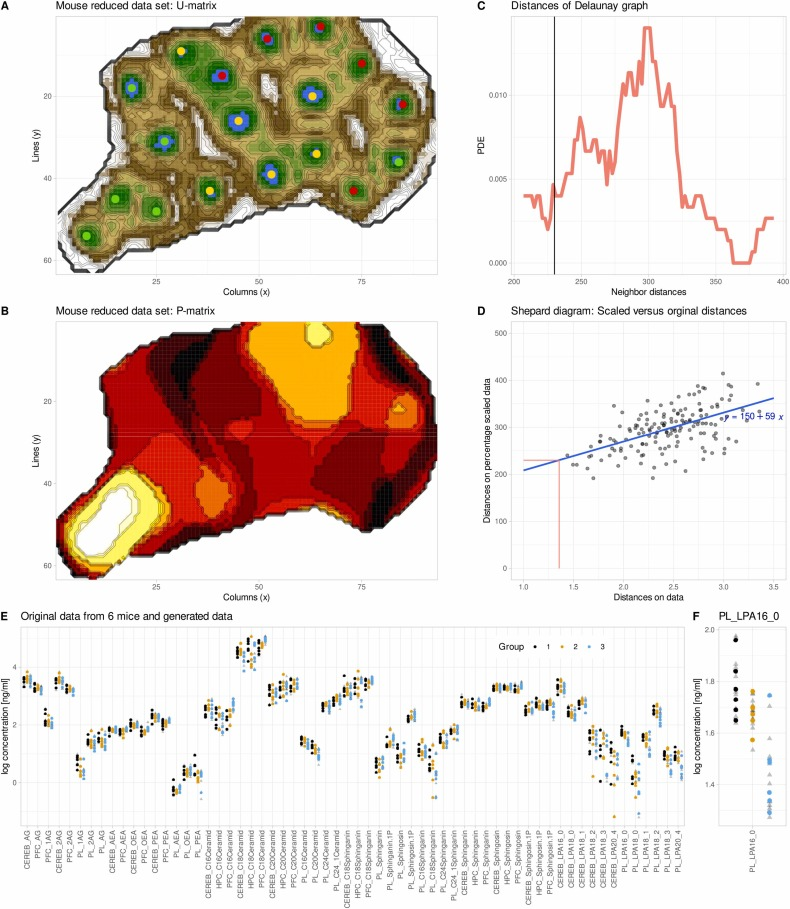
Anschließend nutzte das Forschungsteam genESOM, um die reduzierten Daten zu ergänzen.Nach 20 Trainingsrunden war das Modell immer noch in der Lage, einige Trennungstendenzen der drei Probengruppen im ESOM-Raum zu identifizieren.Dies bestätigt, dass die Daten auch dann noch potenzielle biologische Strukturinformationen enthalten, wenn die statistische Signifikanz verloren geht.
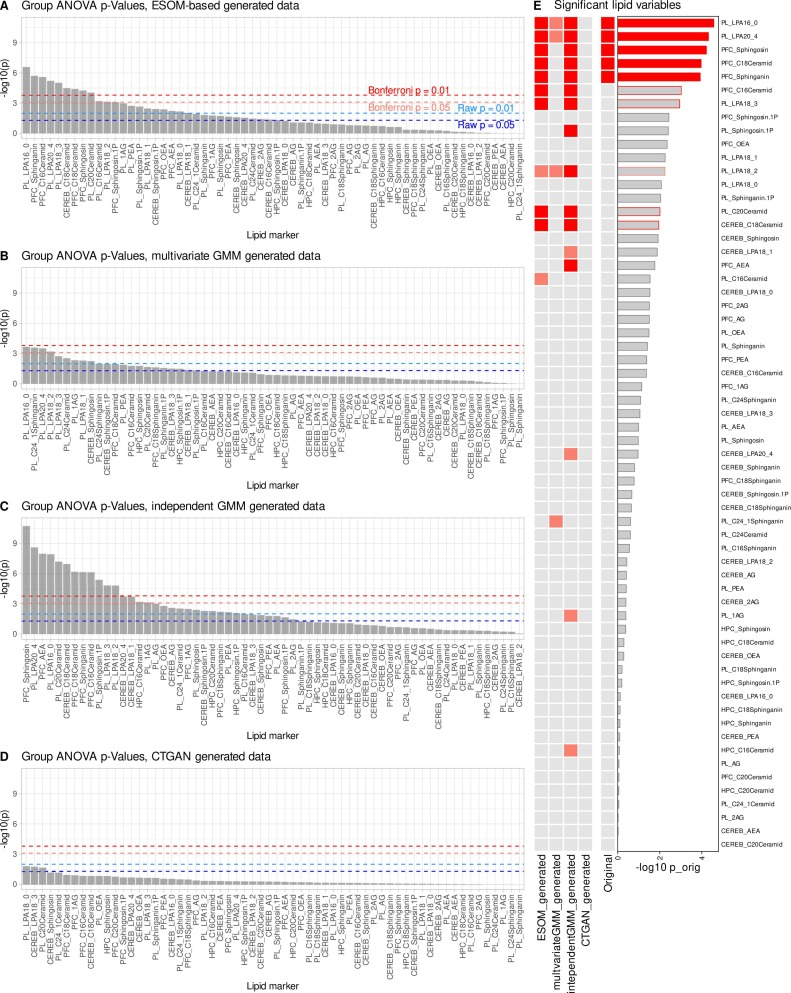
Nach der Datenerweiterung zeigten wichtige Lipidindikatoren wie Lysophosphatidsäure und Sphingolipide im präfrontalen Kortex im Plasma erneut signifikante Unterschiede zwischen den Gruppen. Diese Indikatoren hatten in den Daten mit kleinen Stichproben gänzlich versagt, konnten aber durch die KI-Erweiterung erfolgreich wiederhergestellt werden. Gleichzeitig führte das Modell keine große Anzahl unplausibler neuer Merkmale ein, und es traten lediglich einige zusätzliche Indikatoren mit annähernd gleicher Signifikanz auf.Dies deutet darauf hin, dass genESOM keine neuen Signale aus dem Nichts erzeugt, sondern vielmehr reale biologische Signale verstärkt, die bereits existieren, aber aufgrund unzureichender Stichprobengröße nicht nachgewiesen werden können.
Unter den gleichen Bedingungen mit kleinen Stichproben, wie in der Abbildung unten dargestellt, schnitten die beiden Kontrollgenerierungsmethoden schlecht ab: Das multivariate Gaußsche Mischungsmodell konnte nur einen Teil des ursprünglichen Signals wiederherstellen; das unabhängige Gaußsche Mischungsmodell stellte zwar einige signifikante Indikatoren wieder her, wies aber deutliche falsch-positive Ergebnisse auf; und das bedingte Tabellen-GAN konnte die Kernergebnisse nicht effektiv wiederherstellen und hatte eine hohe Rate falsch-negativer Ergebnisse. InsgesamtgenESOM weist in Szenarien mit kleinen Stichproben eine deutlich bessere Stabilität und Zuverlässigkeit auf als herkömmliche Generierungsmethoden.Es kann wichtige biologische Signale präzise wiedergeben und gleichzeitig Fehlerausbreitung und Fehlinterpretationen streng kontrollieren.
Wie die untenstehende Abbildung zeigt, wurde diese Schlussfolgerung durch eine Analyse des maschinellen Lernens weiter bestätigt: Die verbesserten Daten stellten die Klassifizierungsfähigkeit des Random Forest wieder her, und die ausgewählten Hauptmerkmale stimmten weitgehend mit der ursprünglichen Studie überein.
Letzte Worte
Kleine Stichproben stellen in der biomedizinischen Forschung seit Langem eine Herausforderung dar: Hohe Kosten, ethische Hürden und Schwierigkeiten bei der Probenbeschaffung führen zu unzureichender statistischer Aussagekraft. Traditionelle Datenaugmentation stößt an die Grenzen der Vergleichbarkeit, und generativer KI-Ansatz neigt bei kleinen Stichproben zu falsch-positiven Ergebnissen. Der Durchbruch von genESOM liegt darin, keine Daten zu „erzeugen“, sondern vielmehr bestehende biologische Signale aus begrenzten Daten kontinuierlich zu rekonstruieren.
Das Kerndesign trennt Strukturlernen von Datengenerierung, unterdrückt Fehler durch Dimensionsanpassung und führt eine Negativkontrolle für Echtzeitüberwachung ein. Dadurch entsteht ein zurückhaltender Rahmen, der lediglich Bestehendes verbessert, anstatt Neues zu schaffen. Es ist wichtig zu beachten, dass Verbesserungen reale Experimente nicht ersetzen können; diese Methode befindet sich noch im explorativen Stadium und ihre Anwendbarkeit bedarf weiterer Validierung. Dennoch sendet diese Forschung ein wichtiges Signal: Unter strenger Kontrolle von Fehlern und falsch-positiven Ergebnissen hat generative KI das Potenzial, ein effektives Hilfsmittel für Studien mit kleinen Stichproben zu werden und so zuverlässigere Schlussfolgerungen aus begrenzten Daten zu ermöglichen.








