Command Palette
Search for a command to run...
Wissenschaftler Haben Unabhängig Voneinander Neuartige Materialien Entwickelt, Indem Sie Galliumhaltige Materialien Mithilfe Eines Bayes'schen Optimierungsverfahrens Durch Reverse Engineering analysierten. Die Optimierungsergebnisse Zeichnen Sich Durch Einzigartigkeit Und Neuartigkeit aus.

In der modernen Halbleiterindustrie werden die Grenzen der Materialleistung ständig erweitert. Von hocheffizienten Photovoltaik-Bauelementen über Hochleistungs-Leuchtdioden (LEDs) bis hin zu Hochfrequenzkommunikation und Quanteninformationssystemen – fast alle Schlüsseltechnologien basieren auf einer Kernkompetenz.Präzise Kontrolle über die elektronische Struktur von Materialien, insbesondere die präzise Gestaltung der Bandlücke.Dieses Ziel war im traditionellen System der Materialwissenschaften jedoch lange Zeit schwer zu erreichen.
Der Grund dafür liegt darin, dass die elektronischen Eigenschaften eines Materials nicht allein durch ein einzelnes Element bestimmt werden, sondern von komplexen chemischen Bindungen, der Kristallstruktur, der Elektronenorbitalhybridisierung und den Synergieeffekten mehrerer Elemente beeinflusst werden. Unter den vielen Materialsystemen nehmen galliumbasierte Halbleiter eine Sonderstellung ein.Die hervorragende chemische Vielfalt und die Multivalenzzustandseigenschaften von Gallium ermöglichen es ihm, eine Reihe von einstellbaren elektronischen Eigenschaften aufzuweisen, von einer großen Bandlücke bis hin zu einer kleinen Bandlücke.
Galliumhaltige Verbindungen bilden eine entscheidende Grundlage für wichtige optoelektronische Technologien und Energiewandlungstechnologien wie hocheffiziente Solarzellen, Hochleistungs-LEDs und Hochfrequenz-Kommunikationsgeräte. Sie rücken auch als potenzielle Kandidaten für flexible, biokompatible und implantierbare elektronische Systeme in den Fokus. Trotz jahrzehntelanger Forschung beruht die Entdeckung neuer galliumhaltiger Materialien mit spezifischen elektronischen Eigenschaften jedoch nach wie vor weitgehend auf empirischen Untersuchungen.Dies wird hauptsächlich durch den enormen Gestaltungsspielraum der Komponenten und den hohen Rechenaufwand für Berechnungen auf Basis von Grundprinzipien begrenzt.
Vor diesem Hintergrund hat ein Forschungsteam der Flinders University in Zusammenarbeit mit der Khalifa University in den VAE ein maschinelles Lernverfahren zur Bayes'schen Optimierung (BO) vorgeschlagen, das die Rückentwicklung von Gallium-basierten Bauteilen mit vordefinierten elektronischen Eigenschaften unter Beibehaltung der chemischen Rationalität ermöglicht.
Mithilfe dieses einheitlichen Rahmens,Das System kann autonom neuartige, chemisch wirksame galliumhaltige Materialien erzeugen und eine einstellbare Bandlücke von 0,5–3,5 eV erreichen.Dieser Energiebereich ist von großer Bedeutung für Anwendungen in der Solarenergie, Photonik und Leistungselektronik. Der Bayes'sche Optimierungsprozess kann die Suche adaptiv in den Bereich mit der höchsten gewünschten Verbesserung lenken. Die optimierten Analyseergebnisse zeigen, dass das generierte Material 100% im Vergleich zu den Trainingsdaten einzigartig und neuartig ist und die SMACT-Effektivität im Bandlückenbereich von 1,5–2,5 eV signifikant verbessert wird.
Die zugehörigen Forschungsergebnisse mit dem Titel „Bayesian Optimization-Guided Discovery of Gallium-Containing Semiconductors with Targeted Band Gaps“ wurden in ACS Publications veröffentlicht.
Forschungshighlights:
Das neue Rahmenwerk kann das inverse Materialdesign unter realistischen chemischen Randbedingungen beschleunigen und bietet eine Alternative zu traditionellen Screening-Methoden auf Basis der Dichtefunktionaltheorie (DFT).
* Das neue Framework deckt nicht nur effizient chemisch plausible Bereiche ab, sondern weist im Vergleich zu bestehenden Datenbanken auch einen hohen Grad an Neuheit und Komponentendiversität auf.
* Diese Forschung überwindet die Grenzen der traditionellen statischen Eigenschaftsvorhersage und treibt die Halbleiterforschung hin zu einem datengetriebenen generativen Forschungsparadigma voran.

Papieradresse:
https://pubs.acs.org/doi/10.1021/acsmaterialslett.5c01482
Datensätze: Aufbau eines chemischen Lernraums aus realen Materialdatenbanken
Für diese Studie wurden die Datenbanken NOMAD und Materials Project verwendet, um das Modell zu trainieren.Die Daten umfassen die chemische Zusammensetzung des Materials und den entsprechenden experimentellen Bandlückenwert.Beispiele hierfür sind Ga₄P₄, GaAs, GaN, Ga₂O₃ usw. Der ursprüngliche Datensatz enthält 2.530 Materialzusammensetzungen und deren Bandlückenwerte.
Um die Datenqualität zu gewährleisten, wurden Proben mit fehlenden Werten in den Spalten „Zusammensetzung“ oder „Bandlücke“ entfernt. Nicht-physikalische oder negative Bandlückenwerte wurden ebenfalls eliminiert, und doppelte Datensätze wurden gelöscht. Dadurch verblieben 1.578 gültige Komponenten für die Modellierung. Darüber hinaus wurden die chemischen Formeln mithilfe des pymatgen-Pakets standardisiert, um chemisch äquivalente Terme zusammenzufassen. Die Einheit der Bandlücke wurde einheitlich von Joule in Elektronenvolt (eV) umgerechnet. Im vorverarbeiteten Datensatz lag die Bandlücke zwischen 0,0 und 5,92 eV, mit einem Mittelwert von etwa 1,8 eV und einer Standardabweichung von 1,6 eV.
Die Studie untersuchte zudem die Materialzusammensetzung und berücksichtigte nur Verbindungen mit Elementen aus einem vordefinierten Bereich von Ordnungszahlen, um sicherzustellen, dass sich die Forschung auf galliumbasierte Materialsysteme konzentrierte. Darüber hinaus wurden mehrere weitere Merkmale entwickelt, darunter:
* Anzahl der Elemente in jeder chemischen Formel
* Länge der chemischen Formelkette
* Ein binärer Indikator für das Vorhandensein oder Fehlen von Gallium
Der Datensatz wurde anschließend zufällig im Verhältnis 8:2 in Trainings- und Testdaten aufgeteilt. Die Aufteilung erfolgte auf Komponentenebene, um zu vermeiden, dass chemisch ähnliche Verbindungen gleichzeitig in verschiedenen Datensätzen vorkommen. Zur Bewertung der Robustheit des Modells unter verschiedenen Datenaufteilungsbedingungen wurde zudem eine fünffache Kreuzvalidierung durchgeführt.
Rahmenkonzept: Gemeinsame Entwicklung von maschinellem Lernen und Bayes'scher Optimierung
Diese Studie schlägt ein chemisch beschränktes Bayes'sches Optimierungsmodell (BO) vor.Wie in der Abbildung unten dargestellt, wird zunächst ein Gradient-Boosting-Regressionsmodell verwendet, das auf einem Datensatz von Gallium-basierten Verbundwerkstoffen trainiert wurde, um die Bandlücke des Materials vorherzusagen; anschließend wird eine Bayes'sche Optimierung eingesetzt, um iterativ in einem eingeschränkten Zusammensetzungsraum zu suchen; schließlich werden die generierten Kandidatenmaterialien mithilfe der Werkzeuge SMACT und pymatgen auf chemische Gültigkeit, Neuartigkeit und Einzigartigkeit geprüft, wodurch Gallium-basierte Verbundwerkstoffe mit der besten Leistung identifiziert werden, die bisher noch nicht untersucht wurden.
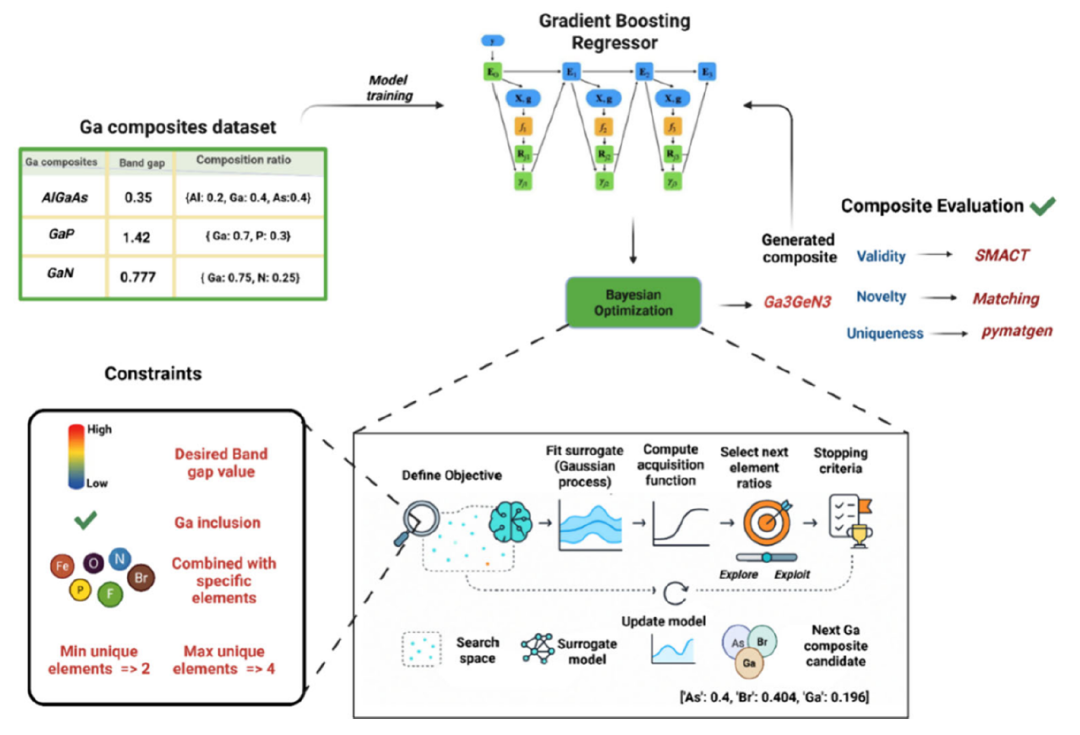
Vorhersagemodellschicht
Diese Studie evaluierte systematisch acht maschinelle Lernalgorithmen für Regression, darunter lineare Modelle, Support-Vektor-Regression, Random Forests, Gradient Boosting und K-Nearest Neighbors (KNN). Die Ergebnisse zeigen, dass das nichtlineare Modell das lineare Modell insgesamt deutlich übertrifft, was auf einen starken nichtlinearen Zusammenhang zwischen Materialzusammensetzung und Bandlücke hindeutet.Das KNN-Modell schnitt am besten ab und erreichte ein R² von 0,812. Auch hinsichtlich der Fehlermetriken übertraf es andere Modelle.
Von allen Kandidatenmodellen wurde schließlich KNN als Ersatzmodell in der Bayes'schen Optimierung ausgewählt.Der Grund dafür ist, dass es über ausgezeichnete lokale Interpolationsfähigkeiten verfügt und auch unter verschiedenen zufälligen Partitionierungsbedingungen eine stabile Leistung beibehält.Im Gegensatz zu baumbasierten Ensemble-Modellen kann KNN Nachbarschaftsbeziehungen im Komponentenmerkmalsraum erhalten, was für die Identifizierung von Ähnlichkeiten zwischen Materialien mit ähnlichen Elementanteilen von entscheidender Bedeutung ist.
In Szenarien der Bayes'schen Optimierung ist diese „lokale Erhaltungsfähigkeit“ besonders wichtig, da Optimierungssuchen sich häufig auf potenzielle Regionen in der Nähe bekannter, qualitativ hochwertiger Kandidaten konzentrieren. Daher können die nichtparametrischen und lokal adaptiven Eigenschaften von KNN dem Optimierer eine gleichmäßigere und zuverlässigere Suchführung bieten und gleichzeitig eine hohe Recheneffizienz in spärlich abgetasteten Materialräumen gewährleisten.
Modul für Bayes'sche Optimierung
Dieser BO-Workflow nutzt das KNN-Surrogatmodell, um die Suche nach galliumhaltigen Komponenten in der Zielbandlücke zu steuern.Durch den Einsatz der Akquisitionsfunktion „Expected Improvement“ wird ein Gleichgewicht zwischen „Exploration“ und „Nutzung“ hergestellt, wodurch Kandidaten für die Stöchiometrie in einem galliumzentrierten Zusammensetzungsraum generiert werden.
Das System legt mehrere Einschränkungen fest, darunter: Jede Komponente darf maximal 4 Elemente enthalten und muss einen Mindestgalliumgehalt aufweisen, um sicherzustellen, dass die Kandidatenmaterialien für galliumbasierte Forschungsthemen relevant bleiben.
Chemisch eingeschränkte Filterschicht
Alle generierten Kandidatenmaterialien müssen mithilfe des SMACT-Tools verifiziert werden, einschließlich Einschränkungen wie Ladungsausgleich, plausibler Oxidationszustand und Elektronegativitätskonsistenz, um sicherzustellen, dass die generierten Materialien nicht nur im mathematischen Raum gültig, sondern auch chemisch realisierbar sind.
Darüber hinaus integriert das Framework Methoden der erklärbaren künstlichen Intelligenz (XAI) und nutzt SHAP zur Analyse der Entscheidungslogik des Modells, wodurch die Materialvorhersage von einer „Black Box“ in ein „erklärbares System“ umgewandelt wird.
Beschleunigung des inversen Materialdesigns unter realistischen chemischen Randbedingungen
Die Forscher entwarfen eine Reihe von Experimenten, um die Leistungsfähigkeit, die strukturellen Merkmale, die Interpretierbarkeit und die chemische Validität des Modells zu bewerten und zu analysieren:
Bewertung der Modellleistung
Hinsichtlich der Bewertung der Modellleistung zeigte das KNN-Modell Stabilität bei der Kreuzvalidierung mit einem R² von etwa 0,60 ± 0,07 und einem RMSE von etwa 1,02 eV, was darauf hindeutet, dass das Modell eine gute Generalisierungsfähigkeit in dünn besetzten chemischen Räumen besitzt.
Wie die nachfolgende Merkmalswichtigkeitsanalyse zeigt, sind Schmelzpunkt, Elektronegativitätsbereich und Elektronegativitätsabweichung Schlüsselfaktoren für die Bandlückenvorhersage. Diese Faktoren stehen in engem Zusammenhang mit der Bindungsstärke und dem Ladungstransferverhalten in Materialien. Mit zunehmender Elektronegativitätsdifferenz verringert sich tendenziell die Bandlücke, während ein Anstieg des Schmelzpunkts und der Kohäsionsenergie mit größeren Bandlücken einhergeht – ein Muster, das mit der klassischen Halbleiterphysik übereinstimmt.
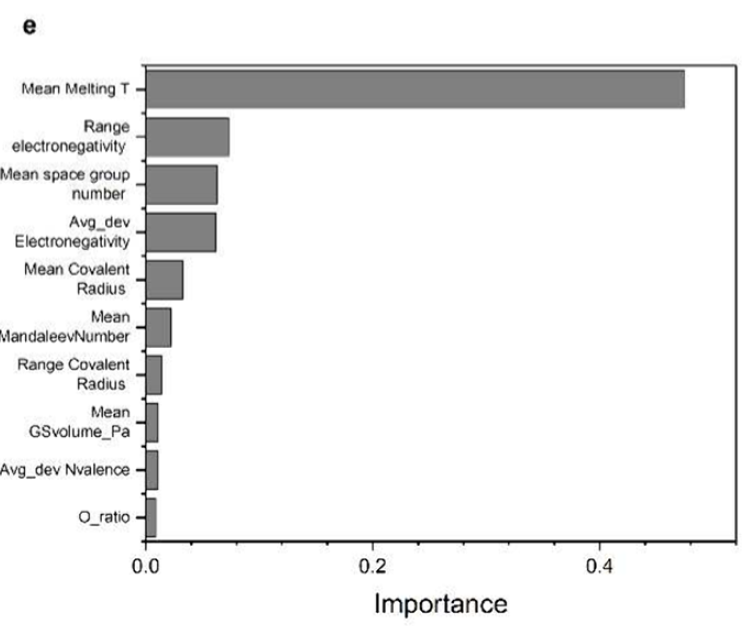
Die Fähigkeit, aus Daten reale chemische Gesetze zu lernen.
Während der Generierungsphase wurden mittels Bayes'scher Optimierung 1.025 Kandidaten für galliumhaltige Komponenten vorgeschlagen, von denen nur 38 das SMACT-Screening bestanden haben, was darauf hindeutet, dass die Anforderungen an die chemische Machbarkeit extrem streng waren.Diese effektiven Materialien konzentrieren sich hauptsächlich im Bereich von 2,0–2,5 eV, was bedeutet, dass sich in diesem Bereich leichter Halbleiter mit mittlerer Bandlücke und sowohl ionischen als auch kovalenten Bindungseigenschaften herstellen lassen. Diese Ergebnisse stimmen sehr gut mit bekannten Systemen wie Ga₂O₃ (≈4,8 eV) und Ga₂S₃ (≈2,5 eV) überein.
Der BO-Suchprozess zeigt auch eine Tendenz zur Clusterbildung in Richtung bekannter galliumhaltiger chemischer Familien (wie Ga–O, Ga–N, Ga–As/Sb) und schlägt neue intermediäre Stöchiometrien in diesen Bereichen vor, wie zum Beispiel: Ga₀.₅₁As₀.₁₆N₀.₂₄Sb₀.₁₀, Ga₀.₁₇₁Sb₀.₁₇₅O₀.₃₆₇F₀.₂₈₆.
Bei Materialien mit großer Bandlücke (>3,0 eV) bevorzugt der Algorithmus sauerstoffreiche Verbindungen, da starke Ga–O-Bindungen die Bandlücke vergrößern. Materialien mit kleinerer Bandlücke (ca. 1,5–2,0 eV) werden typischerweise durch den Ersatz von Sauerstoff durch Schwefel, Selen oder Phosphor erzielt, wodurch stärkere p–p-Wechselwirkungen entstehen. Diese Muster stimmen sehr gut mit bestehenden experimentellen Beobachtungen überein, was darauf hindeutet, dass das Modell reale chemische Gesetze aus den Daten „implizit gelernt“ hat.
Die Fähigkeit, reale „Struktur-Eigenschafts-Beziehungen“ zu erfassen
Um zu bestätigen, dass die erzeugte galliumhaltige Zusammensetzung einem „physikalisch realisierbaren“ Material entspricht, verwendete das Forschungsteam das von Park et al. entwickelte Chemelon-dng-Modell, um dessen Kristallprototyp vorherzusagen, wie in der folgenden Abbildung dargestellt:
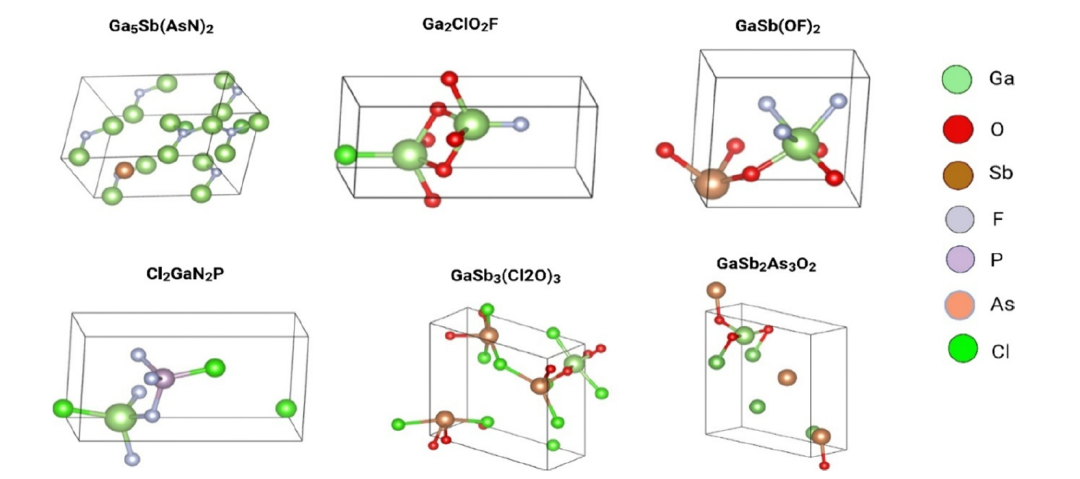
Die mittels SMACT validierten Kandidatenkomponenten zeigten chemisch plausible Koordinationsumgebungen, die von tetraedrischen und oktaedrischen Galliumzentren dominiert wurden. Dies stimmt weitgehend mit bekannten Kristallprototypen wie Ga₂O₃, GaN und GaSe überein. Das Ersatzmodell reproduzierte erfolgreich die empirisch konsistenten Hierarchiebeziehungen der elektronischen Struktur: Oxide: 3,5–4,8 eV, Chalkogenide: 1,8–2,6 eV und Stickstoffverbindungen der Gruppe A: ca. 1,2–2,0 eV, d. h. Bandlücke Oxid > Bandlücke Chalkogenid > Bandlücke Stickstoff der Gruppe A.
Dieses Ergebnis deutet darauf hin, dassDieser Workflow zur Bayes'schen Optimierung ist nun in der Lage, reale Struktur-Eigenschafts-Beziehungen effektiv zu erfassen.
Es ist bemerkenswert, dass keine der 38 verifizierten gültigen Komponenten Duplikate bereits bekannter Materialien waren, was zusätzlich beweist, dass die erzielten Ergebnisse sowohl „Neuheit“ als auch „chemische Konsistenz“ aufweisen.
DFT-Verifizierung
Die Forscher führten anschließend eine DFT-Validierung durch. Die folgende Tabelle fasst die Vergleichsergebnisse der „modellierten Bandlücke“ und der „DFT-berechneten Bandlücke“ der 10 Komponenten zusammen, die die SMACT-Validierung bestanden haben, sowie die entsprechenden Bandlückentypen.
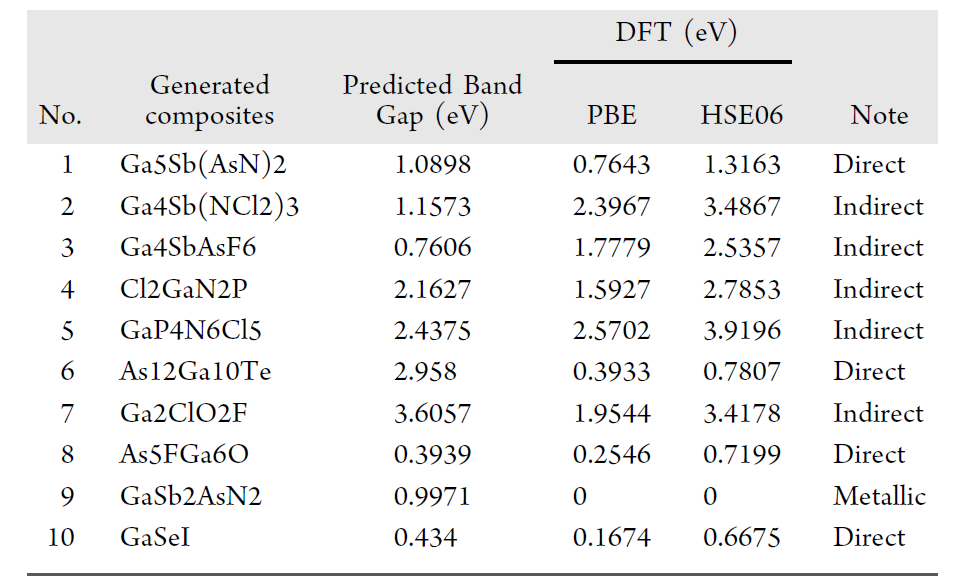
Insgesamt betrug der mittlere absolute Fehler (MAE) 0,890 eV, der mittlere quadratische Fehler (RMSE) 1,158 eV und der Median des absoluten Fehlers 0,784 eV. Trotz gewisser systematischer Abweichungen ist das Verfahren in der frühen Screening-Phase der Materialentwicklung von hohem praktischem Nutzen. Besonders hervorzuheben ist, dass alle validierten Materialien in bekannten Datenbanken nicht zu finden waren, was ihren hohen Neuheitsgrad belegt.
Abschluss
Insgesamt demonstriert diese Studie ein neuartiges Materialdesignparadigma für galliumhaltige Halbleiter: einen automatisierten Generierungspfad von "Daten" zu "neuen Materialien" durch den Synergieeffekt von maschinellem Lernen, Bayes'scher Optimierungssuche und chemischem Constraint-Screening.
Aus industrieller Sicht birgt dieser Ansatz Potenzial für das Design von Photovoltaikmaterialien, die Entwicklung von Leuchtdioden und die Forschung an Halbleitern mit großer Bandlücke. Insbesondere vor dem Hintergrund der rasanten Entwicklung von Leistungselektronik und optoelektronischen Bauelementen der nächsten Generation wächst der Bedarf an Materialien mit steuerbarer Bandlücke rapide, und KI-gestützte Materialdesignmethoden dürften sich als Schlüsselinstrument zur Beschleunigung dieses Prozesses erweisen.
Darüber hinaus beschränkt sich die Bedeutung dieses Rahmens nicht auf das Galliumsystem; seine Methodik lässt sich auch auf Indium, Zinn und sogar bleifreie Halbleitersysteme übertragen und bietet somit einen allgemeinen Ansatz für die rationale Entwicklung komplexer Mehrkomponentenverbindungen. Dies markiert einen Wendepunkt in der Materialwissenschaft: vom erfahrungsbasierten Ausprobieren hin zur algorithmengestützten Materialentwicklung. Künstliche Intelligenz wird dabei zur zentralen Brücke zwischen chemischen Gesetzmäßigkeiten und der Entdeckung neuer Materialien.
Quellen:
https://techxplore.com/news/2026-05-ai-discovery-gen-chips-electronic.html
https://pubs.acs.org/doi/10.1021/acsmaterialslett.5c01482








