Command Palette
Search for a command to run...
Materials AI Bewegt Sich Auf Eine "erklärbare Ära" Zu: Ein Japanisches Team Knackt Die Blackbox Der Hochdimensionalen Spektroskopie Und Identifiziert Schlüsselmerkmale Zur Entdeckung Neuer Materialien.

In den letzten Jahren hat die Anwendung von maschinellem Lernen in der Materialwissenschaft große Aufmerksamkeit erregt. Ihr Anwendungsgebiet hat sich schrittweise von der anfänglichen Vorhersage skalarer Struktur-Eigenschafts-Beziehungen (wie Bandlückenenergie, Punktdefektbildungsenergie, Schmelzpunkt usw.) hin zur Modellierung komplexerer, hochdimensionaler physikalischer Größen erweitert. Eine der größten Herausforderungen stellt die Vorhersage und Analyse von Materialspektren dar.
Spektrale Daten wie dielektrische Funktionen, Spektren (Absorption, Reflexion und Emission) sowie elektronische und phononische Zustandsdichten sind entscheidend für das Verständnis und die Entwicklung von Materialien. Im Vergleich zu skalaren Eigenschaften zeichnen sich hochdimensionale Spektraldaten jedoch durch große Ausgabedimensionen, komplexe Strukturen und starke physikalische Einschränkungen aus. Dies erschwert es traditionellen Methoden des maschinellen Lernens, gleichzeitig Genauigkeit und Interpretierbarkeit zu erreichen. Obwohl Deep-Learning-Modelle Spektren bis zu einem gewissen Grad vorhersagen können, bleibt die mangelnde Interpretierbarkeit ein zentrales Hindernis für ihre weitere Anwendung im Materialdesign.
In diesem ZusammenhangEin Forschungsteam des Tokyo Institute of Science in Japan hat eine Methode zur Interpretation von Deep-Learning-Modellen vorgeschlagen, die hochdimensionale Spektraldaten in der Materialwissenschaft verarbeiten können.Forscher erstellten einen Datensatz mit ab-initio-Berechnungen der optischen Absorptionsspektren von 2681 Oxiden, Chalkogeniden und verwandten Verbindungen. Im Vergleich zu Standard-Dichtefunktionalrechnungen zeigten die berechneten Ergebnisse nach Korrekturen der spektralen Einsatzenergie und Form eine deutlich verbesserte Übereinstimmung mit publizierten experimentellen Spektren.
Die Forscher nutzten den Datensatz und den ALIGNN-Algorithmus auch, um ein hochpräzises Vorhersagemodell für das optische Absorptionsspektrum zu entwickeln.Durch die Kombination von Merkmalsextraktion und Clusteranalyse konnten die Schlüsselelementtypen und ihre Koordinationsumgebungen, die hauptsächlich die Lichtabsorptionsinitiierungsenergie und -intensität bestimmen, erfolgreich extrahiert werden.
Die zugehörigen Forschungsergebnisse mit dem Titel „Deep Learning–Based Extraction of Promising Material Groups and Common Features from High-Dimensional Data: A Case of Optical Spectra of Inorganic Crystals“ wurden in Advanced Intelligent Discovery veröffentlicht.
Forschungshighlights:
* Diese Studie schlägt eine Methode zur Materialklassifizierung durch Merkmalsextraktion und Clusteranalyse hochdimensionaler Spektraldaten vor, wodurch potenzielle Materialgruppen und ihre gemeinsamen Merkmale extrahiert werden.
* Es wird erwartet, dass der in dieser Studie entwickelte Datensatz für Berechnungen auf Basis erster Prinzipien und das maschinelle Lernmodell eine wichtige Rolle bei der zukünftigen Materialforschung und Materialinformatik spielen werden.
Die in dieser Studie vorgestellte Methode ist breit anwendbar und kann zur Klassifizierung und Interpretation verschiedener Spektraldaten eingesetzt werden. Ihre Anwendung beschränkt sich nicht auf die optischen Absorptionsspektren anorganischer Kristalle.

Papieradresse:https://advanced.onlinelibrary.wiley.com/doi/10.1002/aidi.202600007
Datensätze, die mithilfe von Hochdurchsatz-Ab-initio-Berechnungen erstellt wurden
Die Forscher sichteten zunächst Oxide, Chalkogenide und verwandte Materialien aus der Datenbank des Materials Project, die folgende Bedingungen erfüllten: (1) Das Material enthält mindestens eines der Elemente O, S oder Se, und seine Oxidationszahl ist nicht notwendigerweise −2; (2) Das Material enthält nicht die folgenden Elemente: H, Halogene, Edelgase, Mn–Ni, Tc–Rh, Os–Ir, Po, Lanthanide (außer La und Ce) und Actinoide; (3) Das Material weist keine Spinpolarisation auf; (4) Systeme mit der Raumgruppe P1 und/oder mehr als 40 Atomen in der ursprünglichen Einheitszelle wurden aufgrund des hohen Rechenaufwands oder der Unsicherheit in der Kristallstruktur ausgeschlossen.
Die Gesamtzahl der in den Berechnungen nach den Prinzipien der ersten Ordnung verwendeten Materialien betrug 9.808, und die Berechnungsdatenbank wurde gemäß dem in der folgenden Abbildung dargestellten Prozess erstellt.
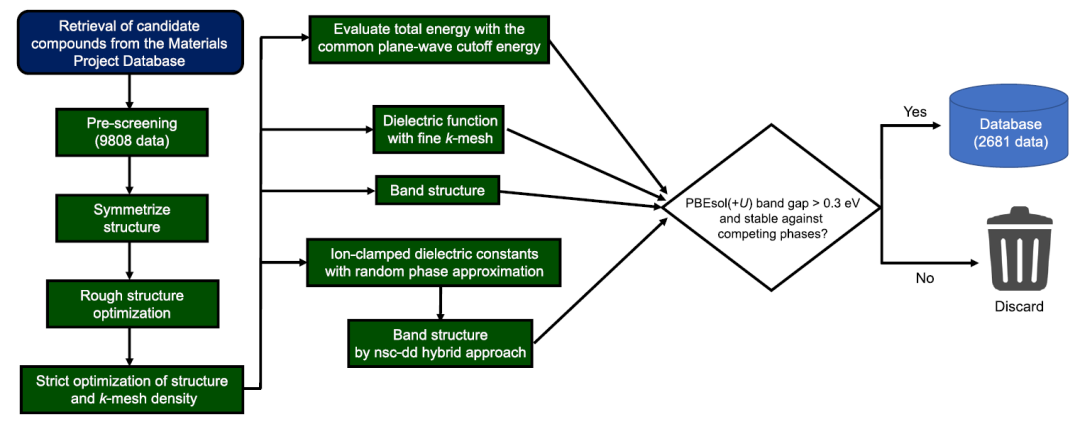
Wie in der Abbildung dargestellt, ist dieser Berechnungsprozess äußerst komplex. Um einen hohen Datendurchsatz bei gleichzeitiger Konsistenz und effizienter Nutzung der Rechenressourcen zu erreichen,Die Forscher nutzten ihr eigenes Programm und stützten sich auf Tools wie pymatgen, FireWorks, Custodian, atomate und vise, um den Prozess zu automatisieren.Alle Ab-initio-Berechnungen wurden mit dem VASP-Softwarepaket durchgeführt. Dieser Arbeitsablauf verwendet PBEsol(+U)-Berechnungen zur Generierung optischer Absorptionsspektren und Verbindungsbildungsenergien sowie nsc-dd-Mischfunktionale und PBEsol(+U)-Berechnungen zur Bestimmung der Bandstruktur.
Bezüglich des Datensatzes für maschinelles Lernen entfernten die Forscher: (1) Materialien, die im Vergleich zur konkurrierenden Phase in der lokalen Datenbank instabil waren; und (2) Materialien mit einer PBEsol(+U)-Bandlücke von weniger als 0,3 eV. Die endgültige Anzahl der verbliebenen Materialien betrug 2681.
Erstellung eines ALIGNN-Modells auf Basis optischer Absorptionsspektren
Konstruktion von Modellen des maschinellen Lernens und Vorhersagegenauigkeit
Auf Modellebene,In dieser Studie wird ALIGNN (Atomistic Line Graph Neural Network) als zentrales Vorhersageframework verwendet, um hochdimensionale optische Absorptionsspektren zu modellieren.Im Vergleich zu herkömmlichen Crystal Graph Convolutional Networks (CGCNN) liegt der Hauptvorteil von ALIGNN in der simultanen Einführung dualer Repräsentationen aus „Atomgraph + Bindungsliniengraph“. Dadurch werden Informationen über Drei-Körper-Winkel explizit kodiert und eine präzisere Darstellung der lokalen Strukturumgebung erreicht. Der obere Teil der folgenden Abbildung zeigt ein schematisches Diagramm der ALIGNN-Architektur.
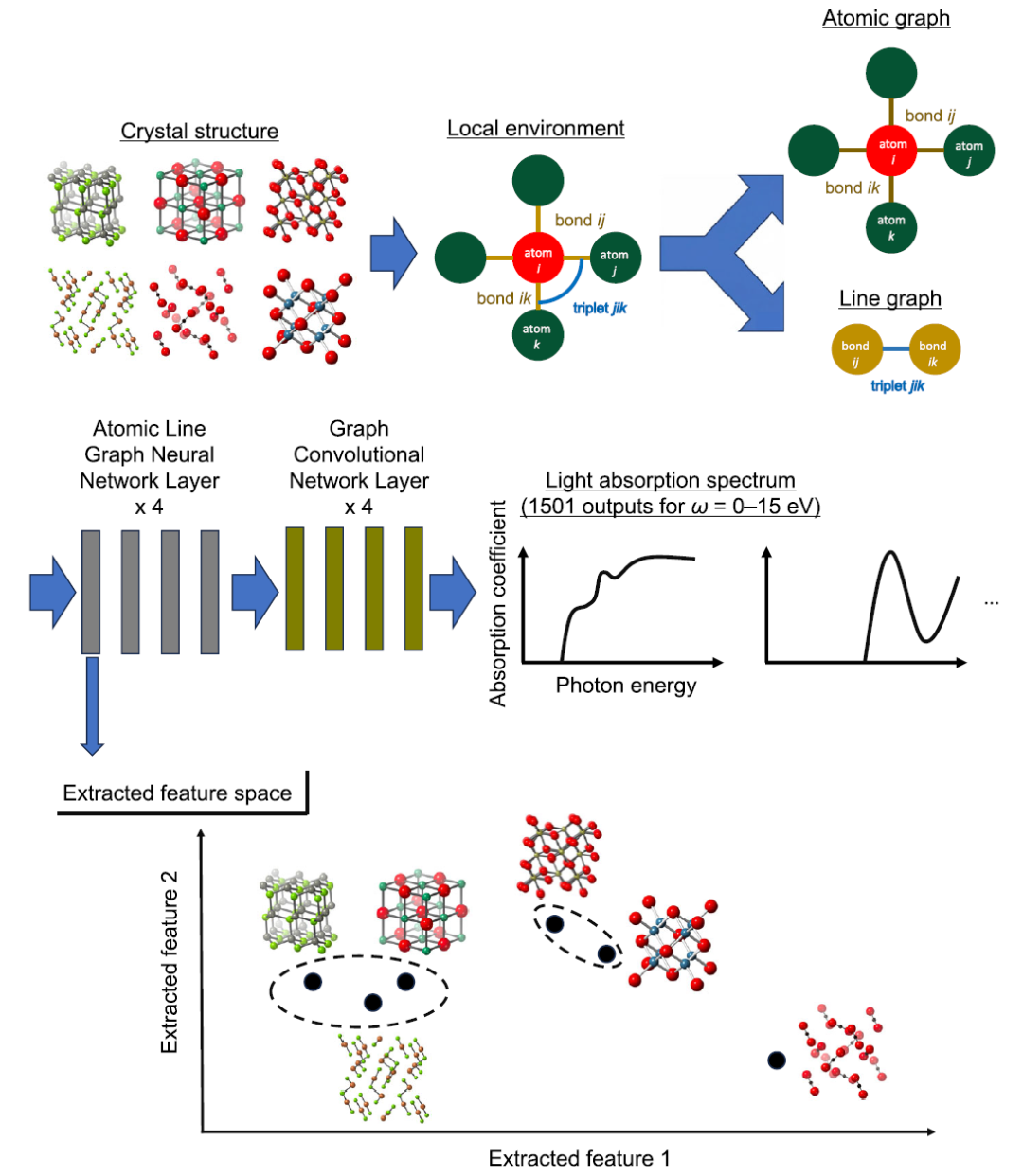
In diesem Rahmen sind Atome Knoten, interatomare Bindungen Kanten und die Beziehungen zwischen den Kanten werden weiter als Liniengraphen konstruiert, wodurch Bindungswinkelinformationen in erlernbare Strukturmerkmale umgewandelt werden.Dieses Design ermöglicht es dem Modell, nicht nur die Abstandsinformationen zwischen zwei Körpern zu erfassen, sondern auch die Wechselwirkung zwischen drei Körpern zu charakterisieren und so das physikalische Verhalten von Kristallen genauer nachzubilden.
Merkmalsextraktion und Clustering
Die Forscher extrahierten Merkmale aus der ersten Schicht des optimierten ALIGNN-Modells und mittelten die Merkmalsvektoren aller Atompositionen für jedes Material, bevor sie eine hierarchische Clusteranalyse durchführten (siehe untere Hälfte der obigen Abbildung). Ziel dieser Methode ist es, Materialien in Gruppen einzuteilen, die Ähnlichkeiten sowohl in den Eingangsmerkmalen (wie der Elementzusammensetzung und atomaren Koordinationsmerkmalen, einschließlich der Anzahl benachbarter Atome, interatomaren Abständen und Bindungswinkeln) als auch in den Ausgangseigenschaften (optischen Absorptionsspektren) aufweisen.
Die Abbildung unten zeigt die optischen Absorptionsspektren der 96 Gruppen, die durch hierarchisches Clustering ermittelt wurden. Die Spektrenformen innerhalb der einzelnen Cluster sind tatsächlich ähnlich, was die Effektivität der Clustering-Methode in dieser Studie bestätigt.

Ergebnisse: Es wurde eine interpretierbare Extraktion der materiellen Populationsstruktur und der physikalischen Mechanismen erreicht.
Um die Fähigkeit des neuen Deep-Learning-Modells zur Verarbeitung hochdimensionaler Spektraldaten in der Materialwissenschaft zu überprüfen, führten die Forscher eine Reihe von Experimenten durch:
Vorhersageleistung
Hinsichtlich der Vorhersageleistung zeigte das ALIGNN-Modell auf dem Testdatensatz insgesamt eine hohe Genauigkeit, wie in der folgenden Abbildung dargestellt.Der mittlere absolute Fehler (MAE) der Vorhersage des Materialabsorptionsspektrums für ungefähr 75% beträgt weniger als 0,14, was darauf hindeutet, dass das Modell komplexe Spektralformen gut reproduzieren kann.
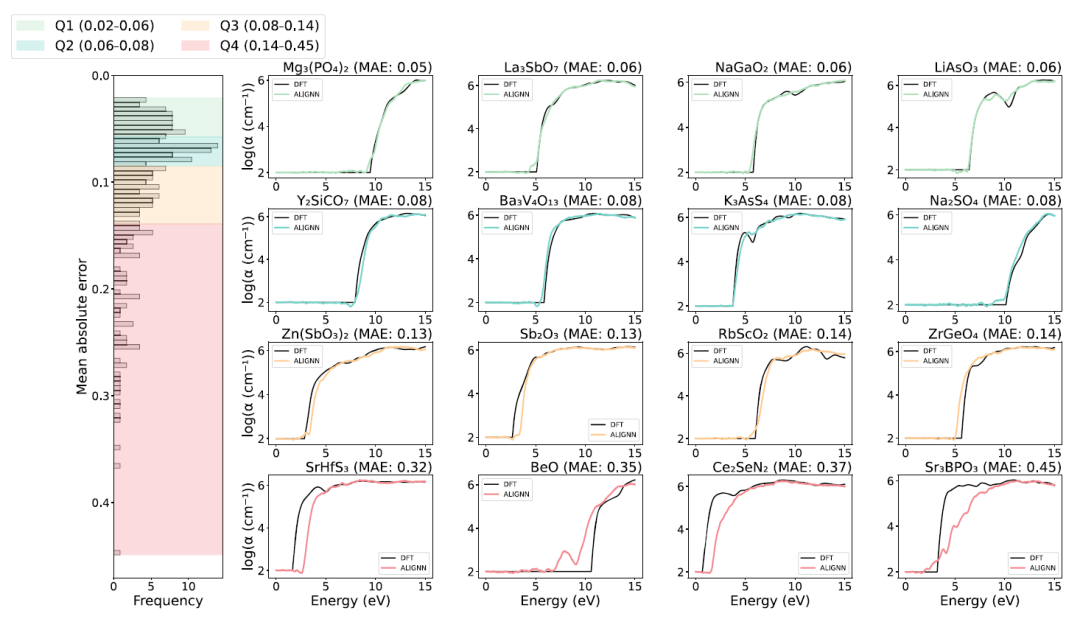
Die rechte Abbildungshälfte zeigt die Vorhersageergebnisse für die vier Materialien mit den größten Fehlern in jedem Quartil. Für die Materialien der ersten drei Quartile stimmen die ALIGNN-Vorhersageergebnisse (farbige Kurven) gut mit den Referenzberechnungen aus ersten Prinzipien (schwarze Kurven) überein. Einige Verbindungen im vierten Quartil weisen jedoch signifikante Abweichungen im Startpunkt ihrer optischen Absorptionsspektren auf. Diese Ausreißerproben zeigen eine geringe Vorhersagegenauigkeit, hauptsächlich aufgrund ihrer einzigartigen elektronischen Strukturen und des Fehlens strukturell ähnlicher Materialien im Trainingsdatensatz.
Die Fähigkeit, die Ausgangsposition von optischen Absorptionsspektren zu erfassen
Obwohl MAE ein globales Maß ist, das den gesamten Spektralbereich abdeckt, untersuchten die Forscher zusätzlich, ob das Modell die lokale spektrale Initiierungsenergie präzise wiedergeben kann. Die folgende Abbildung zeigt ein Paritätsdiagramm: Es vergleicht die niedrigste Photonenenergie, bei der log₁₀ α(ω) in Ab-initio-Berechnungen erstmals den Wert 2,5 überschreitet, mit den ALIGNN-Vorhersagen, wobei α den Absorptionskoeffizienten darstellt.
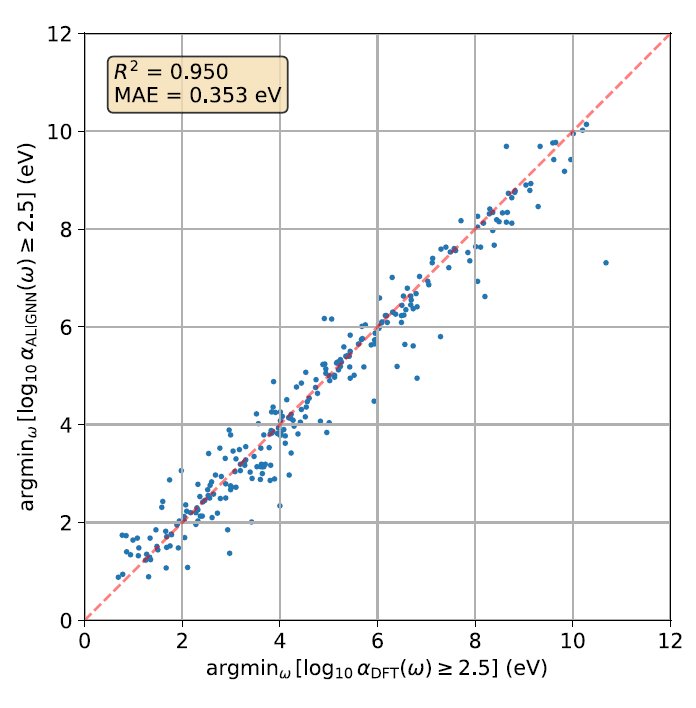
Die Ergebnisse zeigen, dass der vorhergesagte R²-Wert für die Anfangsenergie 0,950 beträgt und der MAE 0,353 eV beträgt, was darauf hindeutet, dass das ALIGNN-Modell die Startposition des optischen Absorptionsspektrums genau erfassen kann.
Interpretierbarkeitsanalyse
Im Rahmen der Interpretierbarkeitsanalyse extrahierten die Forscher Merkmalsdarstellungen aus der ersten Schicht von ALIGNN und führten eine hierarchische Clusteranalyse der Materialien durch, wodurch 96 Materialgruppen entstanden. Die Ergebnisse zeigen, dass…Materialien innerhalb desselben Clusters weisen eine hohe Übereinstimmung in ihrer spektralen Form auf, insbesondere hinsichtlich der Absorptionsbeginnposition und der Steilheit der Absorptionskante, was auf eine signifikante Gemeinsamkeit hindeutet. Dies lässt darauf schließen, dass das Modell in frühen Schichten spektralbezogene Strukturmerkmale erlernt hat.
Weitere Fallstudien zeigen deutliche physikalische Unterschiede zwischen verschiedenen Materialgruppen. So weist beispielsweise Gruppe 74 typischerweise breite Bandlücken und hohe Absorptionskoeffizienten nahe dem spektralen Wendepunkt auf. Abbildung a zeigt, dass alle Materialien dieser Gruppe entweder Vanadium (V) oder Chrom (Cr) enthalten, während die übrigen Kationen hauptsächlich Alkalimetalle sind. Diese Materialien liegen zumeist in Form von VO₄³⁻, CrO₄²⁻ oder Cr₂O₇²⁻ vor, wobei die Kationen tetraedrisch koordiniert sind.
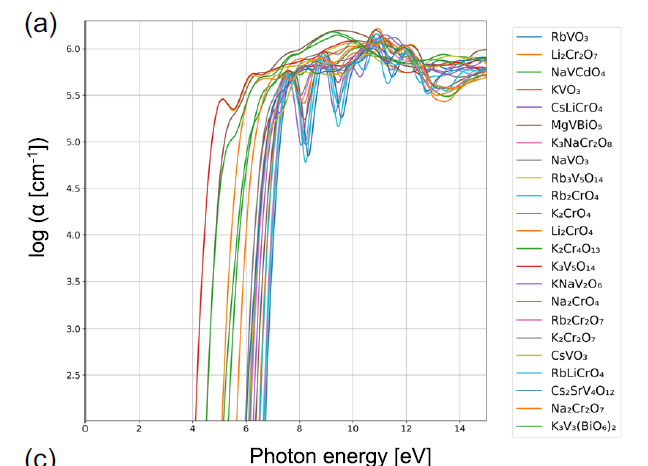
Optische Absorptionsspektren von Substanzen der Gruppe 74, wobei α den Absorptionskoeffizienten darstellt.
Die Forscher nutzten CrystalFingerprintNN, implementiert in Matminer, um den tetraedrischen Koordinationsindex der Kationenplätze in jedem Material innerhalb des Clusters zu berechnen und die Verteilung der Maximalwerte für alle Kationenplätze zu analysieren. Wie in Abbildung b unten dargestellt, weisen die meisten Materialien tatsächlich tetraedrische Koordinationsplätze auf.
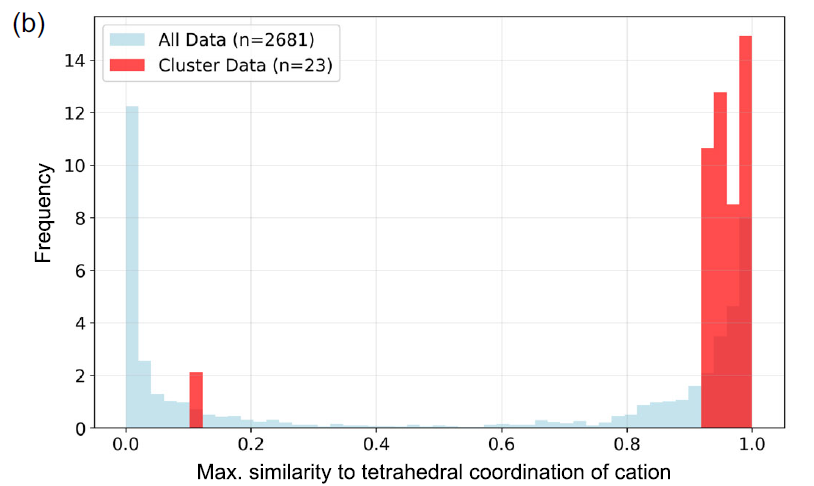
Ähnlichkeitsverteilung der tetraedrischen Koordination zwischen Cluster 74 (rot) und dem gesamten Datensatz (blau).
Aus Sicht der elektronischen Zustandsdichte lässt sich nahe dem Leitungsbandminimum (CBM) ein scharfes Maximum beobachten, das durch Vd- oder Cr-d-Zustände verursacht wird. Die hohen Valenzzustände von V⁵⁺ und Cr⁶⁺ bieten eine große Anzahl unbesetzter elektronischer Zustände, die für optische Übergänge genutzt werden können. Daher ist es aus festkörperchemischer und -physikalischer Sicht plausibel, dass diese Vanadate, Chromate und Dichromate hohe optische Absorptionskoeffizienten aufweisen.
Dieses Verfahren, chemische Mechanismen aus Modellclustering-Ergebnissen abzuleiten, wandelt maschinelle Lernergebnisse von Black-Box-Vorhersagen in eine wertvolle Wissensquelle für die Materialentwicklung um. Die Studie verglich zudem die Ergebnisse des direkten Clusterings auf Basis roher Spektraldaten und stellte fest, dass dieses zwar ähnliche Spektren identifizieren konnte, jedoch Schwierigkeiten hatte, klare chemische Strukturgruppen zu bilden, was zu einer signifikanten Vermischung von Materialtypen führte. Dies unterstreicht den Vorteil des ALIGNN-Merkmalsraums hinsichtlich einer konsistenten Struktur-Eigenschafts-Darstellung.
Abschluss
Die Bedeutung dieser Studie liegt nicht nur in der Entwicklung eines hochpräzisen Vorhersagemodells für die optische Absorptionsspektroskopie, sondern vor allem in der Entwicklung eines methodischen Rahmens, der „Deep Learning Representation Learning“ mit „materialphysikalischer Interpretation“ verbindet. Durch die Kombination des ALIGNN-Modells mit hierarchischer Clusteranalyse gelingt es der Studie, allgemeine Materialgesetze aus hochdimensionalen Spektraldaten zu extrahieren. Dies ermöglicht es Modellen des maschinellen Lernens, nicht nur Ergebnisse vorherzusagen, sondern auch die zugrundeliegende Struktur und die elektronischen Ursprünge dieser Ergebnisse aufzudecken.
Idealerweise sollten die Effekte von Elektron-Loch-Wechselwirkungen, Elektron-Phonon-Kopplung und Punktdefekten berücksichtigt werden, um die spektralen Eigenschaften von Exzitoneneffekten, phononunterstützten elektronischen Übergängen bzw. Defekten zu reproduzieren. Hochdurchsatz-Spektralberechnungen auf Basis von Prinzipien der ersten Ordnung, die diese Effekte einbeziehen, sind jedoch rechentechnisch zu aufwendig, weshalb dies in dieser Studie nicht realisiert werden konnte. Dennoch wird erwartet, dass diese Art von Forschung mit der weiteren Integration präziserer Vielteilchen-Berechnungsmethoden und Modelle des maschinellen Lernens eine zentralere Rolle in der Materialforschung spielen und das Materialdesign von einem erfahrungsbasierten Ansatz zu einer neuen Stufe führen wird, die daten- und mechanismenbasierte Ansätze integriert.