Command Palette
Search for a command to run...
Die Universität Toronto Und Andere Schlugen dnaHNet Vor, Das Die Inferenzgeschwindigkeit Um Das Dreifache Verbessert Und Den Rechenaufwand Für Das Genomlernen Um Fast Das Vierfache reduziert.

Das Genom ist Träger aller genetischen Informationen eines Organismus und bestimmt die Zellfunktion, die individuelle Entwicklung und die Richtung der Artenentwicklung.Die in der Sequenz verborgene "DNA-Syntax" stellt die grundlegenden Regeln des Lebens dar und ist eines der Kernprobleme, die die moderne Biologie dringend lösen muss.Das Verständnis dieser Grammatik steht nicht nur im Zusammenhang mit grundlegenden wissenschaftlichen Kenntnissen, sondern beeinflusst auch direkt die Entwicklung wichtiger Anwendungen wie Krankheitsdiagnostik, Arzneimittelentwicklung und synthetische Biologie.
In den letzten Jahren haben sich auf Basis umfangreicher Sequenzdaten vortrainierte Basismodelle zunehmend als wichtiger Ansatz zur Lösung dieses Problems etabliert. Dank der kontinuierlichen Verbesserung von Rechenleistung, Datenumfang und Modellparametern weisen diese Modelle einen Leistungsanstieg auf, der dem Skalierungsgesetz ähnelt. Modelle wie Nucleotide Transformer und Evo haben ihren Parameterbereich auf Milliarden erweitert und wurden mit Sequenzen verschiedener Spezies trainiert, wodurch signifikante Fortschritte bei Aufgaben wie der Vorhersage von Variationseffekten und der Analyse regulatorischer Elemente erzielt wurden.
DNA-Sequenzen sind jedoch im Wesentlichen kontinuierliche Nukleotidketten mit unscharfen Grenzen – ein grundlegender Unterschied zur natürlichen Sprache. Die beiden derzeit gebräuchlichsten Modellierungsparadigmen sind:Sowohl die Segmentierung auf Basis fester Wörter als auch die Modellierung auf Einzelnukleotidebene stellen einen klaren Zielkonflikt zwischen Ausdrucksstärke und Recheneffizienz dar:Ersteres kann biologische Funktionseinheiten schädigen, während letzteres mit hohem Rechenaufwand verbunden ist. Daher ist die Erzielung eines besseren Gleichgewichts zwischen Rechenaufwand und biologischer Genauigkeit zu einer zentralen Herausforderung geworden. Die dynamische Wortsegmentierung als mögliche Lösung bedarf noch systematischer Erforschung.
In diesem ZusammenhangDas dnaHNet-Modell wurde unter anderem von der Universität Toronto, dem Vector Institute for Artificial Intelligence in Kanada und dem Arc Institute in den Vereinigten Staaten gemeinsam entwickelt.Dies bietet einen neuen Ansatz zur Überwindung der genannten Engpässe. Die zugehörigen Forschungsergebnisse mit dem Titel „dnaHNet: Ein skalierbares und hierarchisches Fundamentmodell für das Lernen genomischer Sequenzen“ wurden als Preprint auf arXiv veröffentlicht.
Forschungshighlights
* Die Recheneffizienz von dnaHNet übertrifft StripedHyena2, und seine Inferenzgeschwindigkeit ist mehr als dreimal so hoch wie die von Transformer.
* Wir schlagen optimale Trainingsstrategien vor, wie z. B. die Festlegung des Kompressionsverhältnisses und den Ausgleich zwischen Encoder und Decoder.
* Erreichte eine Spitzenposition bei Aufgaben ohne Stichproben, wie z. B. der Vorhersage von Variationseffekten und der Klassifizierung der Gennotwendigkeit.
* Es kann kontextabhängige biologische Wortsegmentierung erlernen und sich an funktionelle Regionen wie Codons, Promotoren und intergene Regionen anpassen.

Papieradresse:
https://arxiv.org/abs/2602.10603
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „dnaHNet“, um das vollständige PDF zu erhalten.
Entwurf mehrstufiger genomischer Datensätze für das Training und die Evaluierung von Modellen
Zur Unterstützung des Modelltrainings und der Systemevaluierung wurde in dieser Studie ein mehrschichtiges Datensystem entwickelt.Die Vortrainingsdaten stammen aus einer aufbereiteten Teilmenge der Genome Classification Database (GTDB).Der Prozess folgte strikt den Filter-, Qualitätskontroll- und Redundanzentfernungsverfahren für den Evo-Modell-OpenGenome-Datensatz. Zu den Screening-Kriterien gehörten wichtige Indikatoren wie Assemblierungsintegrität, Kontaminationsgrad und Markergengehalt; nach dem Screening wurde für jeden Spezies-Cluster nur ein repräsentatives Genom beibehalten.
Der endgültige Datensatz umfasst 85.205 Prokaryoten und enthält 17.648.721 Sequenzen.Die Gesamtzahl der Nukleotide beträgt ungefähr 144 Milliarden. Alle Sequenzen wurden aus dem vollständigen Genom extrahiert und in nicht überlappende Segmente von bis zu 8.192 Nukleotiden unterteilt.
Zur Evaluierung erstellten die Forscher einen Testdatensatz aus drei sich ergänzenden Dimensionen, um die Leistungsfähigkeit des Modells umfassend zu untersuchen. ErstensAuf der Ebene der lokalen CodierungseignungZur Bewertung der Fähigkeit des Modells, die lokale Codierungsgrammatik und die Fitnesslandschaft von Proteinen zu charakterisieren, wurden zwölf experimentelle Datensätze auf Nukleotidebene mit insgesamt 21.250 Datenpunkten verwendet, die aus E. coli K12 in MaveDB stammen.
Zweitens,Im Hinblick auf die funktionelle Bewertung auf der Ebene des gesamten GenomsBasierend auf der Essential Gene Database (DEG) wurden binäre Essentialitäts-Tags für 62 Bakterienarten erstellt. Relevante Sequenzen und Annotationen wurden von NCBI bezogen, und eine Sequenzidentität von mehr als 99% mit dem DEG-Eintragsnamen diente als Standard für die Kennzeichnung essentieller Gene. Dies ergab 185.226 Datenpunkte, die zur Bewertung der Fähigkeit des Modells verwendet wurden, Langzeitabhängigkeiten und den genomischen Kontext zu integrieren.
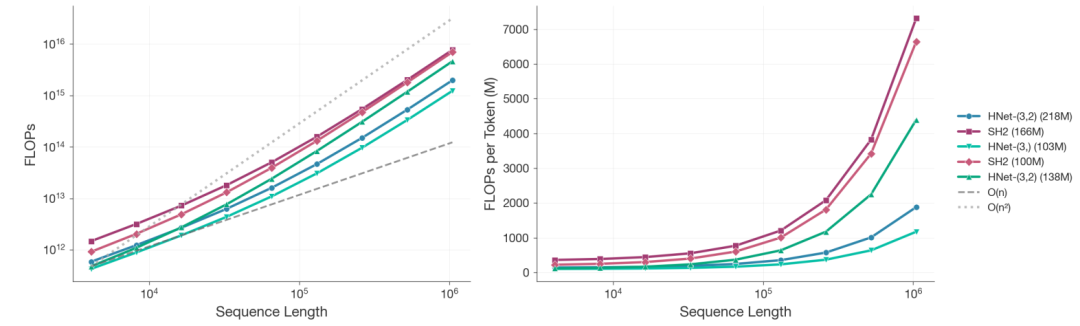
endlich,Im Hinblick auf die strukturelle Interpretierbarkeit,Am Beispiel des Genoms von Bacillus subtilis wird die Sequenz durch Kombination ihrer funktionellen Annotationen in verschiedene funktionelle Regionen unterteilt. Die Fähigkeit zur Strukturmodellierung wird durch die Analyse der Übereinstimmung zwischen den Segmentierungsergebnissen des Modells und den realen biologischen Strukturen verifiziert.
dnaHNet-Modell: Autoregressives Frontier-Modell ohne Wortsegmentierung
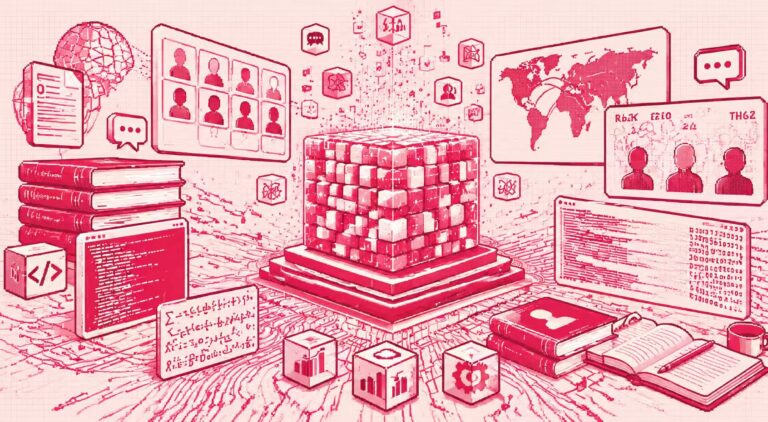
dnaHNet ist ein genombasiertes Modell, das keinen expliziten Segmentierer benötigt.Der Schlüssel liegt im Mechanismus der „dynamischen Segmentierung“, der es dem Modell ermöglicht, die Struktureinheiten in der Sequenz selbstständig zu erlernen.Dieses Design vermeidet die Fragmentierung biologischer Funktionssegmente durch eine Segmentierung mit festen Wörtern und verringert den Rechenaufwand der Nukleotid-für-Nukleotid-Modellierung, wodurch ein besseres Gleichgewicht zwischen Ausdruckskraft und Recheneffizienz erreicht wird.
Hinsichtlich der Modellform,dnaHNet vereint das Genomlernen in einer autoregressiven Sequenzvorhersageaufgabe, die das nächste Nukleotid auf der Grundlage des bestehenden Kontextes vorhersagt.Die Gesamtarchitektur ist hierarchisch aufgebaut, wobei jede Schicht aus einem Encoder, einem Backbone-Netzwerk und einem Decoder besteht: Der Encoder identifiziert mithilfe eines Routing-Mechanismus Stellen in der Sequenz, an denen sich die Information signifikant ändert (z. B. Codongrenzen oder regulatorische Regionen), und komprimiert die Sequenz entsprechend in eine implizite Blockdarstellung; das Backbone-Netzwerk verwendet eine Hybridstruktur, die Mamba und Transformer kombiniert, um die komprimierte Sequenz zu modellieren und dabei sowohl Fernabhängigkeiten als auch wichtige Informationsinteraktionen zu berücksichtigen; der Decoder skaliert die Darstellung dann wieder auf Nukleotidauflösung hoch und gibt die Vorhersageergebnisse aus.
Aufbauend auf dieser Grundlage wurde dnaHNet für Genomdaten mehrfach optimiert. Erstens wurden im Hinblick auf die Parameterzuweisung etwa 301 TP3T Modellkapazität dem Encoder und Decoder zugewiesen, um die Fähigkeit zur Charakterisierung lokaler Strukturen zu verbessern.
Zweitens,Einbeziehung eines zweistufigen, geschichteten Kompressionsdesigns:Die erste Phase konzentriert sich auf die Erfassung kurzskaliger Muster (wie Codons), während die zweite Phase ein breiteres Spektrum funktioneller Strukturen modelliert. Dadurch wird ein Gleichgewicht zwischen Komprimierungseffizienz und Informationsgenauigkeit erreicht. Darüber hinaus integriert der Trainingsprozess autoregressive Vorhersageverluste und Beschränkungen des Komprimierungsverhältnisses, wodurch das Modell die Rechenkosten effektiv kontrollieren und gleichzeitig die Vorhersagegenauigkeit beibehalten kann.
Während der Inferenzphase bestimmt das Modell dynamisch die Blockaufteilungsmethode auf Basis der Randwahrscheinlichkeit, sodass sich die Modellgranularität an den Kontext anpassen und somit der realen Genomstruktur besser entsprechen kann.
dnaHNet reduziert den Rechenaufwand um das 3,89-fache und ist anderen Multitasking-Systemen überlegen.
Um die Leistungsfähigkeit von dnaHNet systematisch zu bewerten, verglich diese Studie es mit zwei gängigen Langsequenz-Genommodellen: StripedHyena2 und Transformer++.Die Experimente umfassen verschiedene Aspekte, darunter Skalierungseigenschaften, Vorhersage des Effekts der Nullstichprobenvariation, Vorhersage der Gennotwendigkeit und Modellierung der biologischen Struktur.
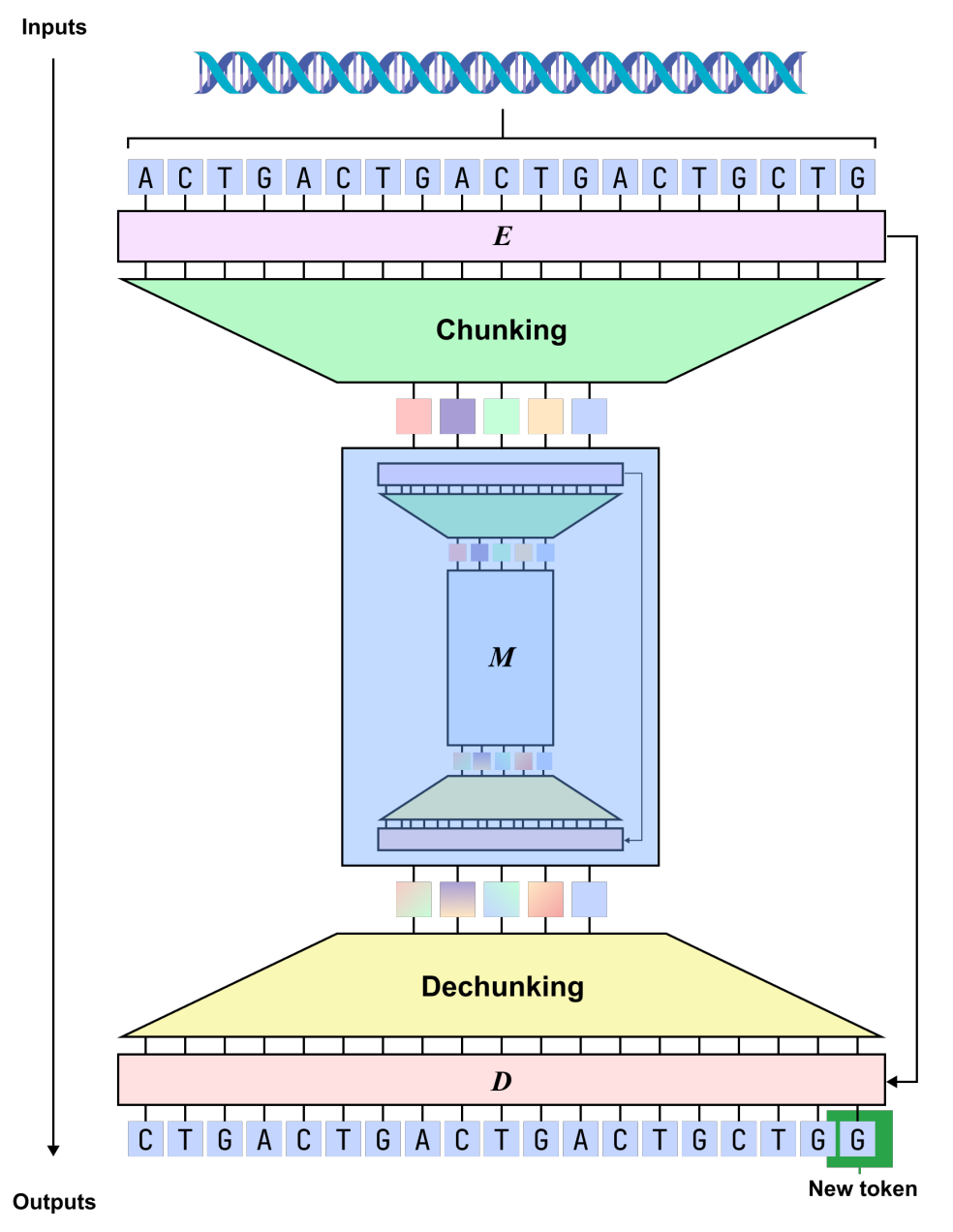
Bei der Skalierungsanalyse,Die Forscher trainierten über 100 Modelle unterschiedlicher Größe mit einem festen Rechenbudget. Als die Sequenzlänge 10⁶ Nukleotide erreichte und die gesamte Rechenleistung 8 × 10¹⁹ FLOPs betrug,Der Rechenaufwand von dnaHNet mit 218 Millionen Parametern ist im Vergleich zu StripedHyena2 mit 166 Millionen Parametern um etwa das 3,89-fache reduziert.Die zweistufige Struktur ist noch effizienter als die einstufige Ausführung.
Die auf der Perplexität basierenden Ergebnisse der Potenzgesetzanpassung zeigen, dassUm das gleiche Leistungsniveau zu erreichen, benötigt StripedHyena2 etwa die 3,75-fache Rechenleistung von dnaHNet.Darüber hinaus weicht die optimale Daten-Parameter-Konfiguration von dnaHNet erheblich vom traditionellen Skalierungsgesetz ab: Bei gleicher Rechenleistung kann die Anzahl der Trainings-Token 140 Milliarden erreichen, während das Vergleichsmodell nur 68 Milliarden aufweist und noch nicht konvergiert ist.
Bei nachgelagerten Aufgaben,dnaHNet übertrifft die Vergleichsmodelle sowohl bei der Vorhersage des Effekts von Proteinvariationen ohne Stichproben (MaveDB) als auch bei der Vorhersage der Gennotwendigkeit (DEG) durchweg.Darüber hinaus erweitern sich seine Vorteile mit zunehmender Rechenleistung. Dies deutet darauf hin, dass sein dynamischer, blockbasierter Mechanismus und seine geschichtete Architektur lokale Codierungssyntax und globale Kontextinformationen effektiver integrieren können, wodurch seine Fähigkeit zur Charakterisierung biologischer Funktionen verbessert wird.
Hinsichtlich der strukturellen Interpretierbarkeit wurde das Genom von Bacillus subtilis mithilfe eines zweistufigen dnaHNet-Modells analysiert. Die Ergebnisse zeigten, dass das Modell spontan biologisch sinnvolle hierarchische Strukturen erlernen konnte: Die erste Stufe reagierte sensitiv auf Codons und konnte Triplettmuster in kodierenden Regionen präzise erfassen; die zweite Stufe konzentrierte sich stärker auf funktionelle Strukturen, wobei Promotoren, Startcodons und intergene Regionen deutlich höhere Segmentierungswahrscheinlichkeiten aufwiesen als kodierende Regionen.
Dieses Ergebnis deutet darauf hin, dassDas Modell verfügt nicht nur über eine hohe Vorhersagekraft, sondern kann auch die funktionelle Organisation des Genoms unter unüberwachten Bedingungen rekonstruieren.Dies bietet einen interpretierbaren Berechnungsweg für die Analyse der „DNA-Grammatik“.
Abschluss
Insgesamt verzichtet dnaHNet auf vordefinierte Sequenzsegmentierungsmethoden und ermöglicht es dem Modell stattdessen, diese automatisch zu erlernen. Experimente zeigen, dass diese dynamische, hierarchische Modellierung nicht nur die Recheneffizienz verbessert, sondern auch die Multiskalenstruktur des Genoms besser widerspiegelt. Langfristig birgt sie, sofern sie aussagekräftige biologische Einheiten stabil erlernen kann, das Potenzial, schwer zu formulierende Muster im Genom aufzudecken und neue Wege für die Forschung in den Bereichen Variationsvorhersage, Funktionserkennung und synthetisches Design zu eröffnen.