Command Palette
Search for a command to run...
Das MIT Hat Das Pichia-CLM-Modell Entwickelt, Um Die „Sprache“ Der Hefe-DNA Zu Erlernen Und so Möglicherweise Die Ausbeute an Exogenen Proteinen Um Bis Zu Das Dreifache Zu steigern.

In der Biopharmazie und der industriellen Biotechnologie ist die effiziente Expression rekombinanter Proteine nach wie vor ein zentraler Faktor für die Produktionskosten und die Durchführbarkeit von Prozessen. Von monoklonalen Antikörpern und Impfstoffantigenen bis hin zu industriellen Enzympräparaten kann selbst eine geringfügige Steigerung der Expressionsrate einen erheblichen wirtschaftlichen Nutzen bringen.
In vielen AusdruckssystemenPichia pastoris (Komagataella phaffii) wird wegen seiner hohen Fermentationskapazität, seines ausgereiften sekretorischen Expressionssystems und seiner hervorragenden Proteinverarbeitungsfähigkeiten geschätzt.Es hat sich zu einem wichtigen Wirt für die industrielle Produktion entwickelt. Ein seit langem bestehendes Problem, das die Branche plagt, ist jedoch, dass selbst bei vollständig identischen Aminosäuresequenzen die bloße Änderung der „synonymen Codons“ in der codierenden DNA zu um Größenordnungen unterschiedlichen Expressionsniveaus führen kann.
Dieses Phänomen beruht auf der Codonverwendungspräferenz (Codon Usage Bias, CUB) – in vielen Organismen werden bestimmte synonyme Codons bevorzugt verwendet. Die Auswahl synonymer Codons beeinflusst die Proteinausbeute, indem sie Transkription, mRNA-Stabilität, Translation, Proteinfaltung, posttranslationale Modifikationen (PTMs) und Löslichkeit beeinflusst.Daher ist die „Codon-Optimierung“ zu einem entscheidenden Schritt bei der Expression exogener Proteine geworden.
Aktuell wurden in der Industrie verschiedene Codon-Optimierungswerkzeuge und -methoden auf Basis von Wirts-CUBs entwickelt, doch diese Methoden führen nicht immer zu hoch exprimierenden Konstrukten. In den letzten Jahren hat sich die künstliche Intelligenz, insbesondere Sequenzmodellierungstechniken, weiterentwickelt.Forscher haben begonnen, Gensequenzen als eine Art "Sprache" zu betrachten und versuchen, die darin enthaltenen impliziten Regeln mithilfe von Methoden zu erlernen, die der Verarbeitung natürlicher Sprache ähneln.
In diesem ZusammenhangEin Forschungsteam des MIT hat ein auf Deep Learning basierendes Sprachmodell namens Pichia-CLM zur Codonoptimierung im industriellen Wirt Pichia pastoris vorgeschlagen, um die Ausbeute an rekombinanten Proteinen zu verbessern.Im Gegensatz zu herkömmlichen Methoden, die auf CUB-Metriken basieren (welche typischerweise nur eine globale Bewertung liefern und den Sequenzkontext ignorieren), nutzt Pichia-CLM Wirtsgenomdaten, um die Aminosäure-zu-Codon-Zuordnung unvoreingenommen zu lernen. Forscher validierten Pichia-CLM experimentell an sechs Proteinklassen unterschiedlicher Komplexität und beobachteten durchweg höhere Expressionsausbeuten im Vergleich zu vier kommerziellen Codon-Optimierungswerkzeugen.
Die zugehörigen Forschungsergebnisse mit dem Titel „Pichia-CLM: Eine sprachmodellbasierte Codon-Optimierungspipeline für Komagataella phaffii“ wurden in PNAS veröffentlicht.
Forschungshighlights:
* Pichia-CLM verwendet Wirtsgenomdaten, um die Aminosäure-zu-Codon-Zuordnung unvoreingenommen zu lernen, wobei nicht nur Wirtspräferenzen, sondern auch Positionsabhängigkeiten und kontextuelle Langzeitbeziehungen berücksichtigt werden.
* Pichia-CLM wurde experimentell an sechs Proteinen unterschiedlicher Komplexität validiert und zeigte durchweg höhere Expressionsausbeuten.
* Die vom Modell erlernten Aminosäure- und Codon-Einbettungen können nach ihren physikochemischen Eigenschaften gruppiert werden, was darauf hindeutet, dass das Sprachmodell physikalisch sinnvolle Muster erfassen kann.
Papieradresse:
https://www.pnas.org/doi/10.1073/pnas.2522052123
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „Pichia pastoris“, um das vollständige PDF zu erhalten.
Erstellung eines umfangreichen Sequenzdatensatzes mit Schwerpunkt auf Pichia pastoris
Anders als bei traditionellen Methoden, die auf empirischen Regeln beruhen, besteht der Kerngedanke von Pichia-CLM darin, Codierungsmuster direkt aus dem Wirtsgenom zu lernen. Zu diesem ZweckDas Forschungsteam erstellte einen umfangreichen Sequenzdatensatz, der sich auf Pichia pastoris konzentrierte.
Um Pichia-CLM zu trainieren, sammelten die Forscher Aminosäure- und Codierungssequenzdaten für zwei Pichia pastoris-Varianten von NCBI: CBS7435 und GS115. Diese wurden durch zuvor in ihrem Labor erhobene Daten ergänzt, darunter Genomsequenzierung und -annotation von GS115, K. phaffii (NRRL Y11430) und K. pastoris.Insgesamt wurden etwa 27.000 Paare von Aminosäure-kodierenden Sequenzdaten verwendet.
Während der Datenverarbeitung tokenisierten die Forscher Aminosäuren und Codons und führten Startsequenzen ein (…). ),Beendigung( ) und füllen ( Der Datensatz ist so beschriftet, dass das Modell Sequenzen unterschiedlicher Länge verarbeiten und Batch-Training unterstützen kann. Zusätzlich ist der Datensatz in Trainings- und Testdatensätze unterteilt, wobei etwa 201 TP3T-Datensätze verwendet wurden, um die Vorhersagefähigkeit des Modells anhand unbekannter Daten zu evaluieren.
Es ist bemerkenswert, dass diese Datenkonstruktionsmethode keine künstlichen „Optimierungsziele“ einführt, sondern vollständig auf natürlichen Genomdaten basiert. Das bedeutet, dass das Modell die tatsächlichen Expressionspräferenzen des Wirts lernt, anstatt künstlich festgelegte Näherungsregeln zu verwenden, wodurch die Grundlage für nachfolgende Leistungsverbesserungen geschaffen wird.
Pichia-CLM verwendet eine GRU-basierte Encoder-Decoder-Architektur.
Modellarchitektur
Der Pichia-CLM verwendet eine Encoder-Decoder-Architektur, die auf Gated Recurrent Units (GRUs) basiert.GRU ist eine verbesserte Architektur rekurrenter neuronaler Netze, die entwickelt wurde, um Langzeit- und Kurzzeitabhängigkeiten in Sequenzdaten zu erfassen. Durch die Steuerung des Informationsflusses mittels Gating-Mechanismen mildert GRU effektiv das bei traditionellen RNNs häufig auftretende Problem verschwindender Gradienten. Darüber hinaus ist die Leistung von GRU mit der von Long Short-Term Memory (LSTM)-Netzen vergleichbar, jedoch benötigt es weniger Parameter und Rechenressourcen und bietet somit in vielen Aufgaben der Sequenzmodellierung deutliche Effizienzvorteile.
Im Vergleich zu einer anderen gängigen Architektur, Transformer, weist GRU eine höhere Recheneffizienz und einen geringeren Ressourcenverbrauch bei kleinen bis mittelgroßen Datensätzen auf.Studien haben gezeigt, dass bei einer Datengröße von etwa 27.000 Sequenzen die Einführung eines Transformers die Komplexität unnötig erhöhen kann, während GRU ein besseres Gleichgewicht zwischen Leistung und Effizienz erzielen kann.
Das Modell verwendet die Aminosäuresequenz eines Proteins als Eingabe und generiert die entsprechende DNA-Sequenz basierend auf Mustern, die aus der Aminosäuresequenz des Wirtsproteins und der codierenden Sequenz gelernt wurden. Die Gesamtarchitektur ist in der folgenden Abbildung dargestellt:
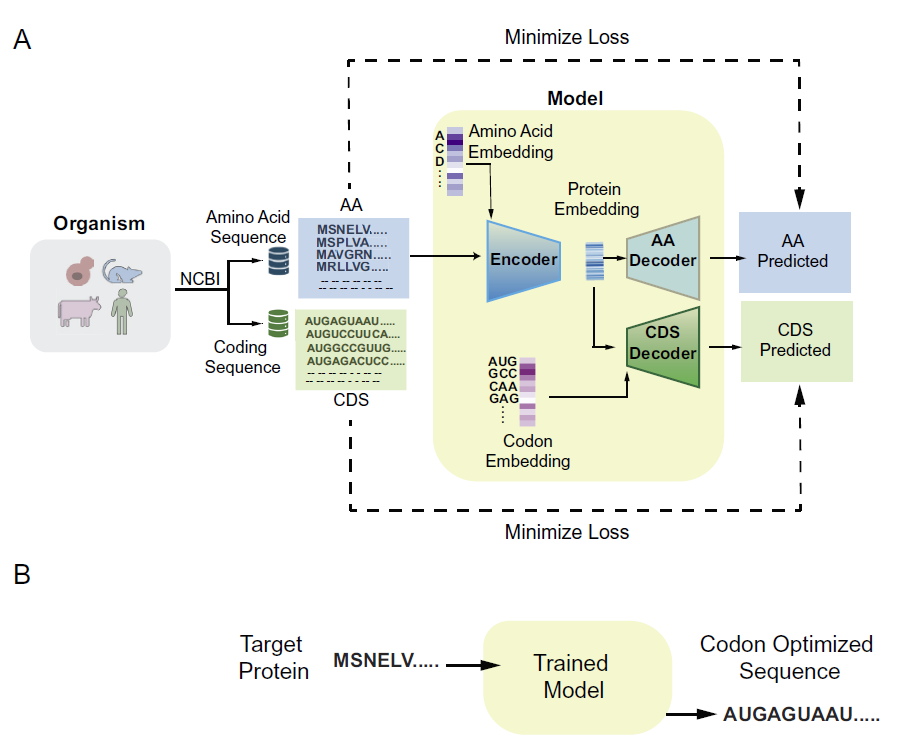
Modelltrainingsprozess
Während des Trainings verwendeten die Forscher einen Validierungsdatensatz (20% des Trainingsdatensatzes) für das Early Stopping, um die Parameter zu optimieren. Gleichzeitig wurde eine Hyperparameter-Auswahl durchgeführt, um den Verlust des Validierungsdatensatzes (Sparse Classification Cross-Entropy) zu minimieren.Die Hyperparameteroptimierung verwendet eine globale Optimierungsstrategie namens Bayes'sche Optimierung, kombiniert mit intern von den Forschern implementiertem Code.
Das Modell umfasst im Einzelnen die folgenden Hyperparameter:
* Aminosäure-Einbettungsdimension
* Codon-Einbettungsdimension
* Anzahl der Einheiten in der Encoderschicht
* Größe der vollständig verbundenen Codon-Schicht im Decoder
* Größe der vollständig verbundenen Aminosäureschicht im Decoder
Während des Modelltrainings ist die Eingabe für den Decoder die tatsächliche kodierte Sequenz (d. h. die realen Codons). In der Vorhersagephase verwendet das Modell das an der vorherigen Position vorhergesagte Codon als Eingabe für die nächste Position und erreicht so eine vollständig autoregressive Vorhersage. Die Sequenzvorhersage endet, sobald ein Stoppcodon gefunden wird.
Nach Abschluss der Architekturauswahl und Validierung der Vorhersagekraft anhand des Testdatensatzes trainierten die Forscher das finale Modell erneut mit dem vollständigen Datensatz und verwendeten weiterhin eine Strategie des vorzeitigen Stoppens, um Überanpassung zu vermeiden. Dieses finale Modell wurde zur Entwicklung der kodierenden Sequenzen exogener Proteine verwendet.
Pichia-CLM kann Konstrukte mit hoher Proteinproduktion erzeugen.
Im Abschnitt zur experimentellen Validierung wählte das Forschungsteam sechs Proteine mit unterschiedlichem Komplexitätsgrad für die Tests aus, darunter:
Menschliches Wachstumshormon (hGH)
* Humaner Granulozyten-Kolonie-stimulierender Faktor (hGCSF)
* VHH Nanobody 3B2 (34)
* Gentechnisch veränderte SARS-CoV-2 RBD-Subunit-Variante (RBD) (35)
* Humanes Serumalbumin (HSA)
* IgG1 monoklonaler Antikörper Trastuzumab (Trast)
Die Leistung von Pichia-CLM bei der Steigerung der Proteinsekretion in Pichia pastoris
Erste,Die Forscher wählten drei vom Menschen stammende Proteine unterschiedlicher Größe und Komplexität aus: hGH, hGCSF und HSA. Anschließend verglichen sie die Unterschiede in der Proteinausbeute (Titer) zwischen Genkonstrukten, die mit Hilfe von Pichia-CLM erzeugt wurden, und ihren nativen codierenden Sequenzen.Insgesamt betrug die Ausbeutesteigerung bei Proteinen wie hGH und hGCSF etwa 25%; bei HSA wurde hingegen ein signifikanter Anstieg um etwa das Dreifache beobachtet.
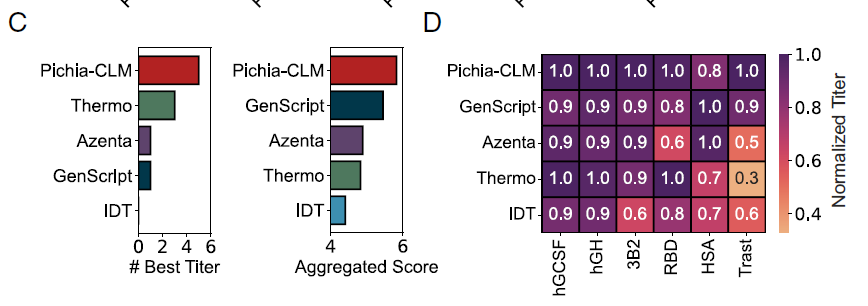
Anschließend verglichen die Forscher Pichia-CLM mit vier kommerziellen Codon-Optimierungstools: Azenta, IDT, GenScript und Thermo Fisher (Thermo). Dabei bewerteten sie sechs Proteine anhand zweier Metriken:
* BestTiter: Die Anzahl der Proteine mit dem höchsten Titer, der mit einer bestimmten Methode erzielt wurde.
* Gesamtscore: Die Summe der relativen Titer verschiedener Proteine (normalisiert auf den Maximalwert).
Gesamt,Pichia-CLM übertraf kommerzielle Algorithmen in beiden Metriken (Abbildung C unten); es erreichte den höchsten Titer bei 5 von 6 Proteinen, wobei es bei HSA aufgrund eines etwas niedrigeren Titers nur zu einem geringfügigen Rückgang des Gesamtscores (ca. 0,2) kam (Abbildung D unten).
Bewertung der genetischen Sequenzmerkmale
Nachdem die Forscher die Leistungsfähigkeit von Pichia-CLM bei der Produktion exogener Proteine validiert hatten, analysierten sie anschließend die genetischen Sequenzmerkmale verschiedener entworfener Konstrukte.Wie auch andere beschriebene Protein-Sprachmodelle stützt sich die Codon-Optimierung typischerweise auf eine oder mehrere Metriken zur Codon-Nutzungspräferenz (CUB) für Design oder Evaluierung. Daher wurden in dieser Studie Daten von sechs Testproteinen verwendet, um die Korrelation zwischen diesen CUB-Metriken und der Proteinausbeute zu untersuchen.
Die Ergebnisse zeigten, dass keiner dieser Indikatoren eine durchgängig hohe Korrelation mit der Ausbeute über verschiedene Proteine hinweg aufwies. Beispielsweise betrug im Fall von HSA (siehe Abbildung A unten) die maximale positive Korrelation mit der Codonvolatilität und der Codonfrequenzverteilung (CFD) lediglich 0,43, während die maximale negative Korrelation mit dem Codonpaar-Score (CPS) nur 0,25 betrug.
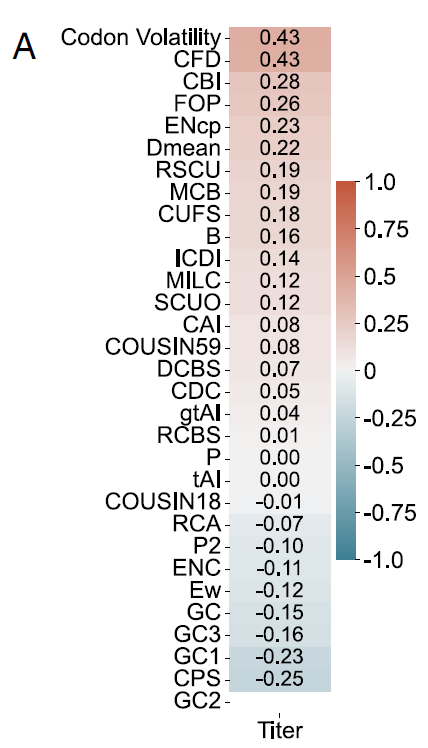
Globale CUB-Metriken, die auf Basis der gesamten Sequenz berechnet werden, weisen erhebliche Einschränkungen bei der Charakterisierung von Merkmalen im Zusammenhang mit der exogenen Proteinproduktion auf.Dies deutet auf die Notwendigkeit neuer Bewertungsmetriken zur Beurteilung von Codon-Optimierungswerkzeugen hin, kombiniert mit einer strengen experimentellen Validierung verschiedener Proteine – ein Ergebnis, das die theoretische Grundlage der traditionellen Codon-Optimierung direkt in Frage stellt.
Sequenzmerkmalsauswertung
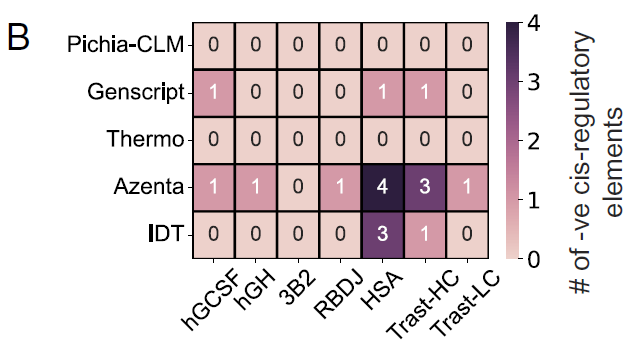
Die Forscher untersuchten auch das Vorhandensein negativer cis-regulatorischer Elemente in verschiedenen codon-optimierten Konstrukten, die die regulatorischen Mechanismen des Wirts beeinträchtigen könnten und daher in exogenen DNA-Sequenzen möglichst vermieden werden sollten.
Von den 6 getesteten ProteinenIn den mit Pichia-CLM entworfenen Konstrukten wurden keine negativen cis-regulatorischen Elemente nachgewiesen; im Gegensatz dazu enthielt GenScript in drei von sechs Proteinen ein negatives cis-regulatorisches Element; Azenta und IDT erzeugten Sequenzen, die in mindestens einem Protein drei bis vier solcher Elemente enthielten.Wie in Abbildung B dargestellt:
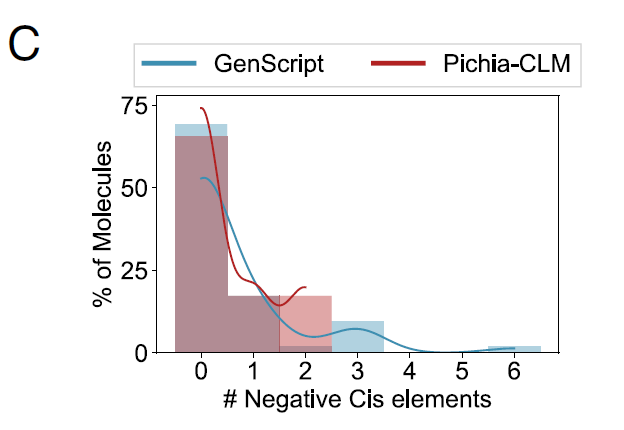
Die Forscher analysierten außerdem die Leistung von Pichia-CLM bei 52 biotechnologiebezogenen Proteinen, und die Ergebnisse zeigten:Die Proteinsequenz von 75% enthält überhaupt keine negativen cis-regulatorischen Elemente, während die verbleibenden 25% höchstens zwei solcher Elemente enthalten.Im Gegensatz dazu erzeugte der leistungsstärkste kommerzielle Algorithmus, GenScript, immer noch Konstrukte mit 3 bis 6 negativen cis-regulatorischen Elementen in Proteinen von ungefähr 15%, wie in Abbildung C unten dargestellt:
Zusammenfassend zeigen diese Ergebnisse, dass Pichia-CLM nicht nur Proteinkonstrukte mit hoher Ausbeute erzeugen kann, sondern auch wichtige genetische Sequenzmerkmale erlernt und ein Gleichgewicht zwischen mehreren Faktoren herstellt, wodurch robuste Codierungssequenzen entworfen werden, die für die Expression im Wirt geeignet sind.
KI beschleunigt die Industrialisierung der Proteinproduktion
In der biopharmazeutischen Industrie ist die Steigerung der Proteinproduktionseffizienz seit jeher ein Schlüsselfaktor für den Erfolg von Forschung und Entwicklung, deren Umsetzung in die Praxis und die Kommerzialisierung. Von monoklonalen Antikörpern über rekombinante Impfstoffe bis hin zu verschiedenen Fusionsproteinen und Enzympräparaten wächst die Marktnachfrage stetig, und die Anforderungen an Ausbeute, Stabilität und Konsistenz steigen kontinuierlich.
Um dieses Ziel zu erreichen, hat die Industrie ein mehrschichtiges Optimierungssystem entwickelt: Auf der Ebene der Wirtsorganismen haben sich neben den traditionellen E. coli und Saccharomyces cerevisiae auch Pichia pastoris und Säugetierzellen aufgrund ihrer überlegenen posttranslationalen Modifikationsmöglichkeiten und Expressionseffizienz zu den wichtigsten Produktionsplattformen entwickelt; auf der Ebene des Moleküldesigns umfasst dies neben der Codonoptimierung die Regulierung der Promotorstärke, das Screening von Signalpeptiden, das mRNA-Struktur-Engineering sowie die Optimierung der Proteinfaltung und des Sekretionswegs; und auf der Prozessebene spielen die Hochdichtefermentation, die Optimierung der Zufuhrstrategie und die Kontrolle der Bioreaktorparameter ebenfalls eine entscheidende Rolle für die Endausbeute.
Außerhalb dieses Systems,Eine neue Art von „Dezellularisierungstechnologie“ entwickelt sich rasant: die zellfreie Proteinsynthese (CFPS).Diese Technologie umgeht das Zellwachstum und nutzt direkt das Transkriptions- und Translationssystem in Zelllysaten, um eine schnelle Proteinexpression zu erzielen. Sie findet breite Anwendung in der Entwicklung und Produktion von Antikörpern, Enzymen und sogar Antikörper-Wirkstoff-Konjugaten. Das zellfreie Proteinsynthesesystem (CFPS) selbst ist jedoch ein hochkomplexes, multivariates System mit Dutzenden von Komponenten wie DNA-Templates, Enzymsystemen, Energiedonatoren, Aminosäuren und ionischen Umgebungen. Der Kombinationsraum ist extrem groß, und traditionelle, erfahrungsbasierte Optimierungsmethoden erzielen oft kein optimales Verhältnis zwischen Kosten und Ausbeute.
Vor diesem Hintergrund erweist sich die KI-gestützte automatisierte Optimierung als äußerst vielversprechend. Kürzlich veröffentlichte OpenAI in Zusammenarbeit mit dem führenden Unternehmen für synthetische Biologie, Ginkgo Bioworks, bahnbrechende Forschungsergebnisse.Das auf dem großen Sprachmodell GPT-5 basierende „geschlossene Automatisierungssystem“ hat eine doppelte Optimierung der zellfreien Proteinsynthese (CFPS)-Technologie erreicht – die Gesamtproduktionskosten der Technologie wurden um 401 TP3T gesenkt, die Reagenzienkosten signifikant um 571 TP3T reduziert und der Proteinsynthesetiter um 271 TP3T verbessert.
Zukünftig werden ähnliche Ansätze auf ein breiteres Spektrum biotechnologischer Produktionsszenarien ausgeweitet. Von der Optimierung von Stoffwechselwegen in Zellfabriken über die Echtzeitsteuerung von Fermentationsprozessen bis hin zum intelligenten Design von Expressionskonstrukten – künstliche Intelligenz wird schrittweise in alle Aspekte der Proteinmedikamentenproduktion integriert.
Quellen:
1.https://www.pnas.org/doi/10.1073/pnas.2522052123
2.https://phys.org/news/2026-02-ai-yeast-dna-language-boost.html#google_vignette
3.https://mp.weixin.qq.com/s/Qkl6j9HcFB7W_Y5Xh-9BCw