Command Palette
Search for a command to run...
KI-gestützte Neuentwicklung Verschiedener Niedermolekularer Bindungsproteine: Ein Südkoreanisches Team Entdeckte Ein Protein, Das Stresshormone Selektiv Erkennen kann.

In den Lebenswissenschaften und der synthetischen Biologie stellt die Entwicklung von niedermolekularen Bindungsproteinen mit hoher Affinität und Spezifität seit jeher eine zentrale Herausforderung für die Realisierung von Biosensoren und molekularen Schaltern dar. Bisher basierte dieser Ansatz hauptsächlich auf dem Screening und der Modifizierung natürlicher Proteine oder auf physikalischen Modellentwürfen, die auf bestehenden Proteinstrukturen beruhen. Dies schränkte jedoch stets die Vielseitigkeit und Skalierbarkeit ein.
In Anbetracht dessenEin Forschungsteam der Abteilung für Biowissenschaften am Korea Advanced Institute of Science and Technology (KAIST) hat mithilfe von Deep Learning-gestützten Methoden zur Generierung von Proteinstrukturen und zum Sequenzdesign verschiedene niedermolekulare Bindungsproteine von Grund auf neu entwickelt, wobei die NTF2-ähnliche Faltung als zentrales „universelles Rückgrat“ diente.Darüber hinaus entwickelten sie daraus einen Sensor, der der chemisch induzierten Dimerisierung (CID) ähnelt. Forschern gelang es, ein Protein zu entwerfen, das selektiv das Stresshormon Cortisol erkennt, und darauf basierend einen KI-Biosensor zu entwickeln. Dieser Fall geht über das Proteindesign hinaus und ebnet den Weg für eine praktisch messbare Sensortechnologie. Er löst die seit Langem bestehende Herausforderung der Erkennung kleiner Moleküle im Bereich des Proteindesigns.
Die zugehörigen Forschungsergebnisse mit dem Titel „Small-molecule binding and sensing with a designed protein family“ wurden in Nature Communications veröffentlicht.
Forschungshighlights:
* Nutzung künstlicher Intelligenz zur Entwicklung von Proteinen von Grund auf (de novo) und deren Anwendung in funktionalen Biosensoren.
Bei traditionellen Methoden geht es vor allem darum, natürliche Proteine zu finden oder bestimmte Funktionen zu modifizieren, während in dieser Studie künstliche Intelligenz zur Entwicklung von Proteinen mit den gewünschten Funktionen eingesetzt wird.
Die Forschungsergebnisse können in Bereichen wie Krankheitsdiagnostik, Entwicklung neuer Medikamente und Umweltüberwachung breit angewendet werden.

Papieradresse:
https://www.nature.com/articles/s41467-026-70953-8
Weitere Artikel zu den Grenzen der KI:
https://hyper.ai/papers
Datensatz: Aufbau des NTF2-Backbones
Um die Designziele zu erreichen,Die Forscher erzeugten zunächst einen Satz von NTF2-Strukturen (Satz 1: 1.615 Rückgrate) mithilfe einer „Halluzinationsmethode“ auf Familienebene und verwendeten dann ProteinMPNN, um die Sequenzen dieser Rückgrate neu zu gestalten.Der AlphaFold-Algorithmus wurde verwendet, um Proteine zu screenen, die sich in die entworfenen Strukturen falten konnten (Satz 2: 3.230 Rückgrate). Darüber hinaus wurde die Rosetta-Parametrisierung zur Generierung der Rückgratstruktur und ProteinMPNN für das Sequenzdesign verwendet, gefolgt von AlphaFold zur Strukturvalidierung (Satz 3: 6.838 Rückgrate), wie in der folgenden Abbildung dargestellt:
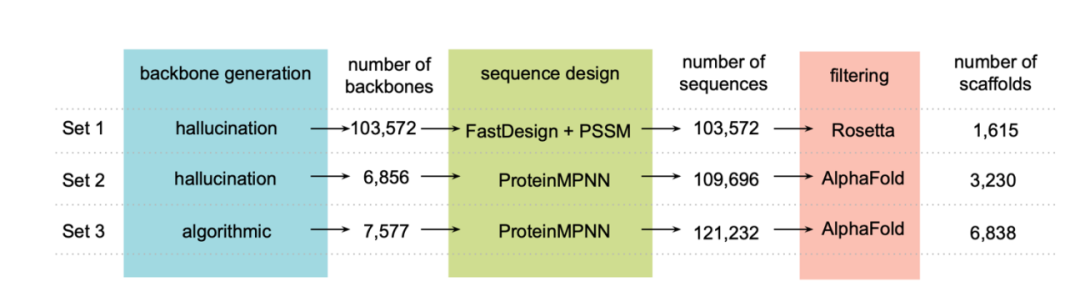
Nach dem Screening erhielten die Forscher schließlich auch die kodierenden Oligonukleotide zur experimentellen Charakterisierung, darunter: 630 HCY-bindende Proteine, 1.661 ROC-bindende Proteine, 16.276 WRF-bindende Proteine, 9.024 APX-bindende Proteine, 19.390 IRI-bindende Proteine und 7.573 OHP-bindende Proteine.
Entwicklung der NTF2-Proteinfamilie mit unterschiedlichen Taschengeometrien
Die NTF2-Faltung besteht aus drei α-Helices und einem gekrümmten 6-strängigen β-Faltblatt. Diese Strukturen bilden zusammen die große interne Verbindungstasche, die für diese Faltungsfamilie charakteristisch ist, wie in der folgenden Abbildung dargestellt:
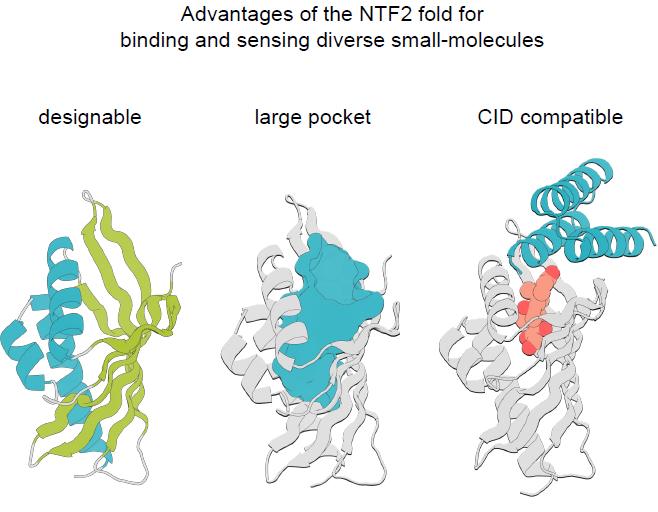
Die Vielfalt dieser Faltung in der Natur rührt hauptsächlich von den langen und unregelmäßigen Ringen und der einzigartigen Quartärstruktur her, die beide die Geometrie und Funktion der kombinierten Tasche beeinflussen.Ziel dieser Studie ist es, eine Familie von NTF2-Proteinen mit unterschiedlichen Taschengeometrien zu entwerfen, die eine breite Palette kleiner Moleküle aufnehmen können, während gleichzeitig Schleifenregionen minimiert werden, um ihre Modularität und Designbarkeit zu erhalten.Der gesamte Designprozess ist im folgenden Diagramm dargestellt:
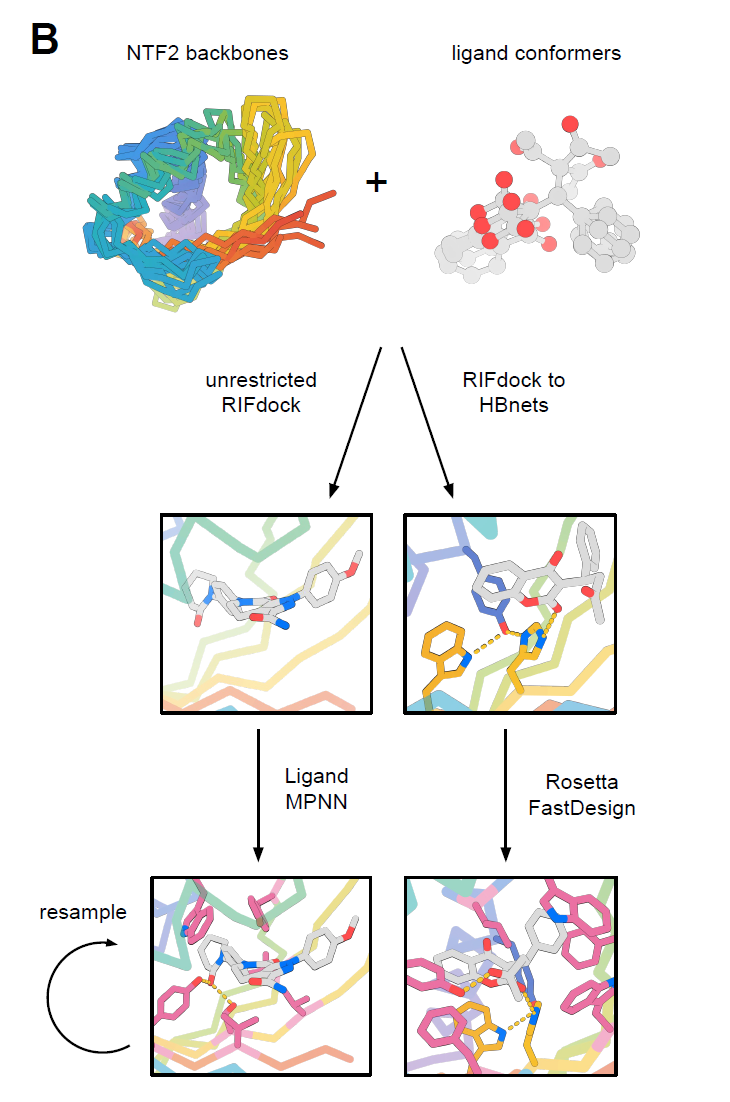
Nachdem über 10.000 NTF2-basierte Proteine mit unterschiedlichen Taschengeometrien generiert worden waren, nutzten die Forscher RIFdock, um sechs kleine Moleküle mit verschiedenen chemischen Eigenschaften und Strukturen in die zentralen Taschen dieser Proteine einzufügen. Zu diesen kleinen Molekülen gehörten das Stresshormon Cortisol (HCY), das Antikoagulans Warfarin (WRF), das Muskelrelaxans Rocuroniumbromid (ROC), das Antikoagulans Apixaban (APX), das aus dem Krebsmedikament Irinotecan abgeleitete, tumorhemmende Molekül SN-38 (IRI) und das Hormon 17-α-Hydroxyprogesteron (OHP).
Die Konstruktion polarer Grenzflächen stellt eine bedeutende Herausforderung im Proteindesign dar, insbesondere bei Proteinen, die kleine Moleküle binden. Dies erfordert die Einführung polarer Aminosäurereste in die innere Bindungstasche, um mit den polaren funktionellen Gruppen der Liganden zu interagieren und gleichzeitig die Gesamtstabilität des Proteins zu erhalten. Um dieses Problem zu lösen, verfolgten die Forscher zwei Strategien:
Methode 1 (RIFdock zu HBNets)
Die Forscher integrierten HCY, WRF, ROC, APX und IRI in das 1-Grundgerüst und forderten mindestens eine Protein-Kleinmolekül-Interaktion, die durch HBNet-Reste vermittelt wird. Anschließend optimierten sie das Design mithilfe eines Rosetta-Designs, das auf natürlichen Sequenzen basiert und eine positionsabhängige Bewertungsmatrix verwendet, die von Proteinen der NTF2-Familie abgeleitet wurde, um das Sequenzdesign zu steuern.
Methode 2 (unbeschränktes RIFdock)
OHP, APX und IRI wurden mithilfe von unbeschränktem RIFdock in die Backbones der Sets 2 und 3 eingefügt, und das Sequenzdesign erfolgte mit LigandMPNN. LigandMPNN ist eine Variante von ProteinMPNN, die speziell auf Protein-Kleinmolekül-Komplexdaten trainiert wurde und die explizite Berücksichtigung der Ligandenpräsenz während des Designprozesses ermöglicht.
Bei der Überprüfung der Design-Ergebnisse verwendeten die Forscher Rosetta, um die Anzahl der Wasserstoffbrückenbindungen zwischen Protein und Ligand, die Bindungsenergie (ddG) und die Kontaktmoleküloberfläche (CMS) zu berechnen; für Methode 2 kombinierten sie außerdem Einzelsequenz-AlphaFold-Vorhersageergebnisse, um Designs zu überprüfen, die gleichzeitig die Zielfaltungsstruktur und die Bindungsstelle reproduzieren konnten (siehe Abbildung unten).
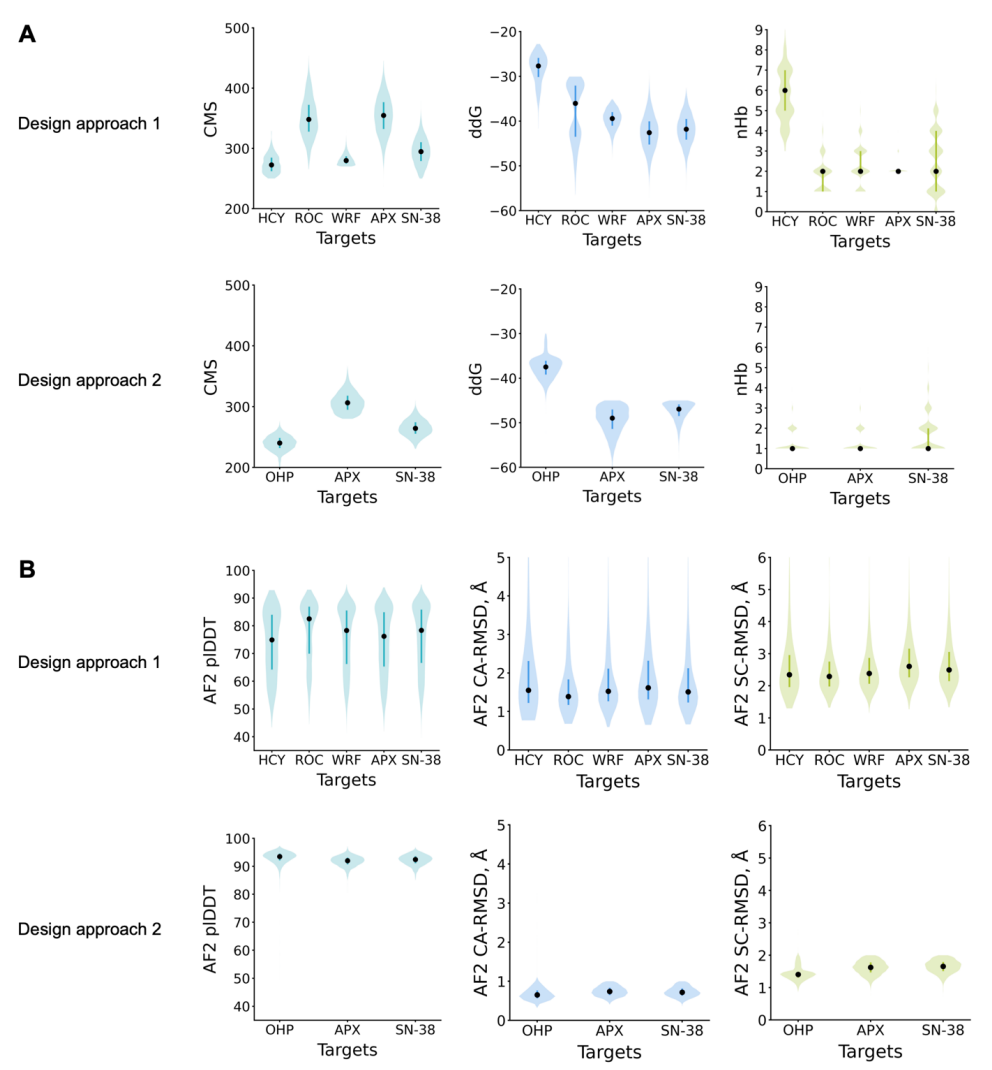
Ergebnispräsentation: NTF2-basierte niedermolekulare Bindungsproteine können in Biosensoren eingesetzt werden.
Die Forscher entwarfen eine Reihe von Experimenten, um die Wirksamkeit der in dieser Studie vorgeschlagenen Designstrategie zu überprüfen:
Design und strukturelle Charakterisierung von Bindungsproteinen
Um die Genauigkeit der entworfenen niedermolekularen Bindungsproteine zu überprüfen, wurden die Kristallstrukturen zweier Protein-Ligand-Komplexe aufgeklärt: des Cortisol-bindenden Proteins hcy129 und des Apixaban-bindenden Proteins apx1049. Insbesondere wurde hcy129 mittels ProteinMPNN einer Oberflächenremodellierung unterzogen, um die Kristallinität zu verbessern. Dadurch konnte die hochauflösende Struktur mit einer Auflösung von 1,5 Å im Cortisol-Komplex erfolgreich bestimmt werden. Die Strukturausrichtung zeigte eine hohe Übereinstimmung der Faltung mit dem Designmodell, mit einem Cα-RMSD von 1,1 Å (Abbildung A unten). Auch die wichtigsten Wasserstoffbrückenbindungsreste und Ligandenkonformationen stimmten präzise überein (Abbildung B unten), was darauf hindeutet, dass das vorab erstellte Wasserstoffbrückennetzwerk (HBNet) die präzise Gestaltung polarer Wechselwirkungen ermöglichte.
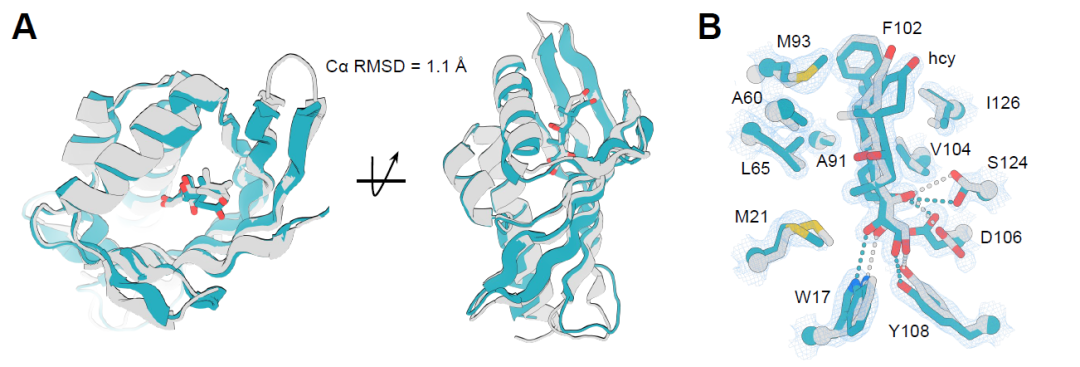
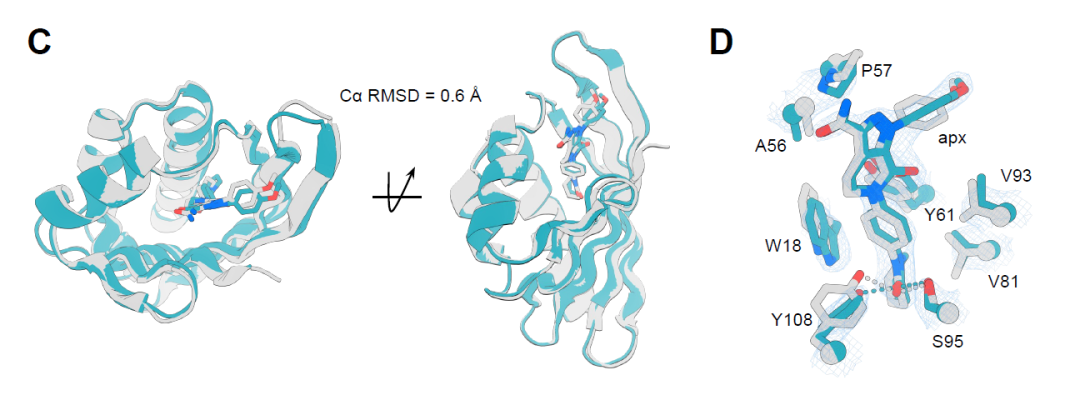
Die Kristallstruktur des Apx1049-Apixaban-Komplexes weist hingegen eine Auflösung von 2,1 Å auf und zeigt damit eine höhere Übereinstimmung mit dem Designmodell. Der Cα-RMSD-Wert beträgt lediglich 0,6 Å über einen Bereich von 113 Aminosäureresten (Abbildung C unten). Die Protein-Ligand-Wechselwirkungen bilden das Design nahezu perfekt ab, einschließlich wichtiger Wasserstoffbrückenbindungen und π-π-Wechselwirkungen zwischen aromatischen Resten (Abbildung D unten). Dadurch wird die Ligandenkonformation stabilisiert und eine hochgradig formkomplementäre Bindungstasche gebildet. Diese Ergebnisse belegen, dass mit dieser Designstrategie eine hochpräzise Konstruktion der Protein-Ligand-Grenzfläche auf atomarer Ebene erreicht wird.
Entwurf einer Spezifitätsbewertung für Bindungsproteine
Um die Spezifität der entwickelten Proteine zu bewerten, wurden in dieser Studie systematisch sechs Bindungsproteine mit sechs Liganden getestet. Albumin, das unspezifische Bindungseigenschaften aufweist, diente als Kontrolle. Die Ergebnisse zeigten, dass hochaffine Proteine wie hcy129.1, iri807.1 und apx1049 eine gute Spezifität bei der Bindung an ihre jeweiligen Zielstrukturen aufwiesen, während Albumin an die meisten Liganden nahezu keine Bindung zeigte. Dies bestätigte die Effektivität der Designstrategie.
Darüber hinaus ist im Warfarin (WRF)-System die Bindungsaffinität von Albumin (KD ca. 5,0 μM) zu diesem ähnlich derjenigen des designten Proteins wrf1071 (KD ca. 1,1 μM), was darauf hindeutet, dass die unspezifische Bindung an stark hydrophobe Liganden weiterhin eine Herausforderung darstellt.
Insgesamt wurde mit dieser Methode ein gewisser Grad an hoher Spezifität bei der Erkennung erreicht, es besteht jedoch noch Raum für weitere Optimierungen bei der Unterscheidung strukturell ähnlicher Moleküle und der Verbesserung der Selektivität für hydrophobe Liganden.
Biosensorkonstruktion (Design und Charakterisierung von Cortisol-induzierten Heterodimeren)
Cortisol ist in physiologischen Proben typischerweise in niedrigen nanomolaren Konzentrationen vorhanden. Plasma-Cortisolwerte über 38 nM können jedoch als diagnostisches Kriterium für Erkrankungen wie das Cushing-Syndrom dienen. Um die Bindungsaffinität von hcy129 zu Cortisol für die Biosensorik zu verbessern, erstellten Forscher eine kombinatorische Mutantenbibliothek basierend auf günstigen Mutationen, die in Einzelpunkt-Sättigungsmutations-Experimenten (SSM) identifiziert wurden. Das Screening erfolgte mittels Hefe-Display, und es wurde eine signifikante Steigerung der Bindungsaffinität beobachtet (siehe Abbildung unten).
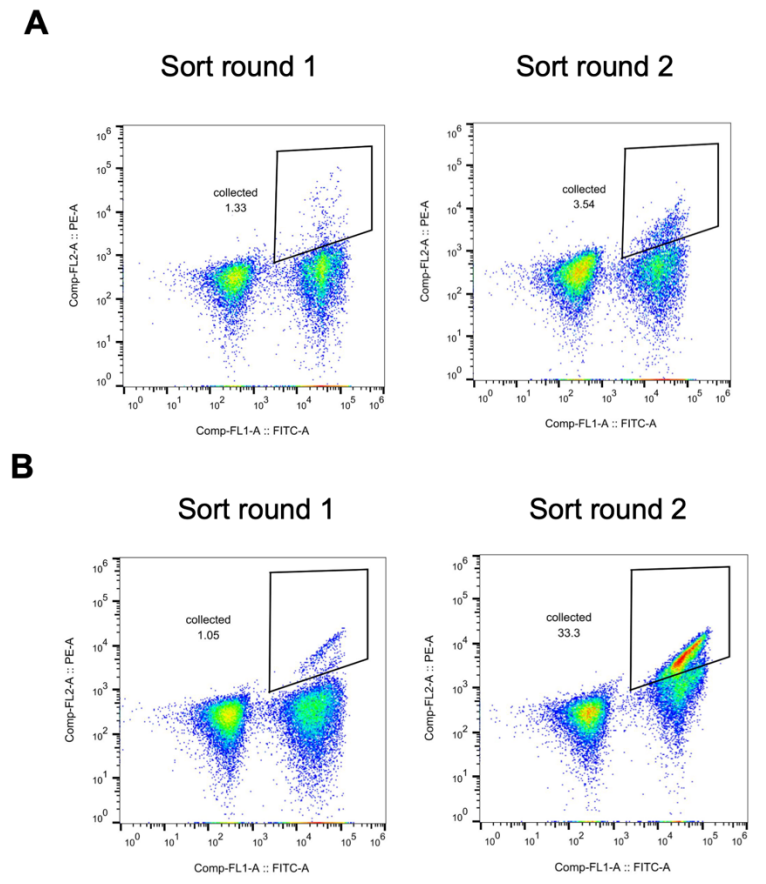
Anschließend wählten die Forscher die optimale Variante aus der Bibliothek aus, exprimierten sie in E. coli und charakterisierten sie mittels isothermer Titrationskalorimetrie (ITC). Die Dissoziationskonstante (KD) dieser Variante hcy129.1 betrug 68 nM und war damit 31-mal höher als die des ursprünglichen Designs (Abbildung C unten). Die Strukturanalyse zeigte, dass die erhöhte Affinität hauptsächlich auf einer stärkeren hydrophoben Wechselwirkung mit Cortisol beruhte (Abbildung D unten).
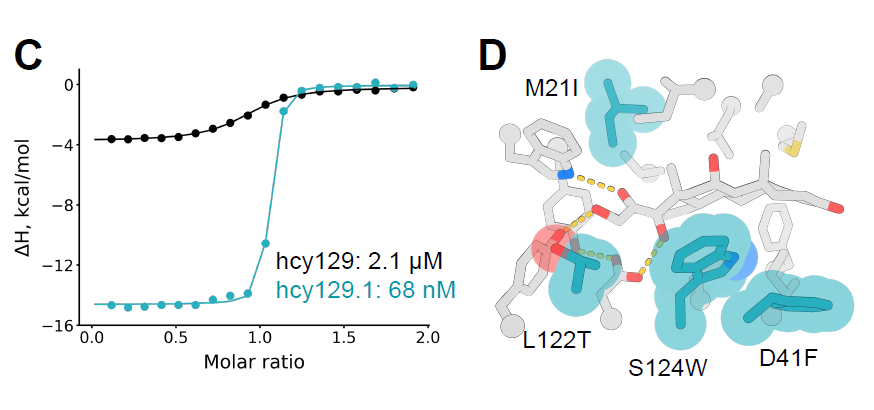
Aufbauend auf dieser Grundlage wurde in der Studie ein Cortisol-abhängiges Heterodimersystem entwickelt. Durch Modifizierung der Struktur von hcy129.1 und Einführung eines kleinen Proteingerüsts wurden computergestütztes Design und Screening mit Methoden wie RIFdock, Rosetta und ProteinMPNN durchgeführt. Schließlich wurde ein kleines Protein, miniH11, erhalten, das einen ternären Komplex mit hcy129.1 und Cortisol bilden kann.
Experimente zeigten, dass das System nur in Gegenwart von Cortisol einen stabilen Komplex bildet. Darüber hinaus wurde das System mit einem NanoBiT-Luciferase-System fusioniert, um Cortisol nachzuweisen. Dabei wurde eine EC50 von ca. 72 nM (siehe Abbildung H unten) ermittelt, was mit der Bindungsaffinität übereinstimmt und somit die Effektivität des Designs bestätigt. Gleichzeitig sank die Affinität des Systems in Abwesenheit von Cortisol signifikant, was auf eine starke Ligandenabhängigkeit der Dimerisierung hindeutet.
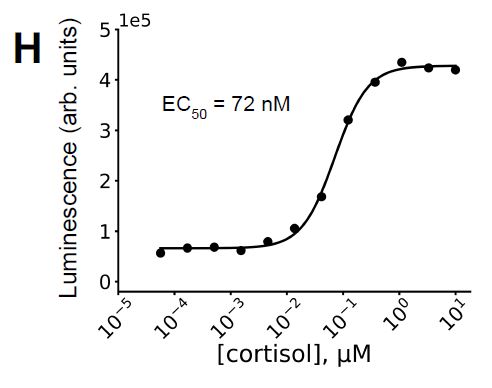
Im äquimolaren (200 nM) hcy129.1_CID-SmBiT- und miniH11-LgBiT-System wurden die Cortisol-abhängigen Lumineszenz-Antwortkurven untersucht...
Gesamt,Diese Arbeit zeigt, dass auf NTF2 basierende niedermolekulare Bindungsproteine zu funktionalen Biosensoren weiterentwickelt werden können.
Abschluss
Insgesamt bietet diese Studie einen neuen Weg für das De-novo-Design von niedermolekularen Bindungsproteinen: Durch die Verwendung von Modellen der künstlichen Intelligenz zur präzisen Charakterisierung von Protein-Ligand-Wechselwirkungen auf atomarer Ebene wird ein Wandel von der "Entdeckung oder Modifizierung natürlicher Proteine" zur "Anpassung funktionaler Proteine nach Bedarf" erreicht und eine effektive experimentelle Validierung abgeschlossen.
Dies stellt nicht nur einen Quantensprung in der Proteinentwicklung dar, sondern erweitert auch deren Anwendungsgebiete erheblich – von der präzisen Detektion von Biomarkern in der Früherkennung von Krankheiten über die gezielte molekulare Erkennung in der Arzneimittelentwicklung bis hin zur Echtzeit-Erfassung von Schadstoffen im Umweltmonitoring. Mit zunehmender Reife dieser Technologie werden hochspezifische und programmierbare, maßgeschneiderte Biosensoren voraussichtlich eine entscheidende Brücke zwischen den Lebenswissenschaften und praktischen Anwendungen schlagen.
Quellen:
https://www.nature.com/articles/s41467-026-70953-8
https://phys.org/news/2026-04-ai-proteins-built-specific-compounds.html