Command Palette
Search for a command to run...
ICLR 2026 | NVIDIA/Universität Oxford Und Andere Schlagen Eine Methode Zur Generierung Von Proteinbindern Auf Atomarer Ebene Mit Modernster Leistung (SOTA) vor.

Im Bereich der Computerbiologie,Die Entwicklung von Proteinen, die präzise an spezifische Zielstrukturen binden können, ist eines der wichtigsten und anspruchsvollsten Probleme.Sie steht nicht nur in direktem Zusammenhang mit Schlüsselbereichen wie der Arzneimittelentwicklung, der Biotherapie und dem Enzym-Engineering, sondern bestimmt auch die Obergrenze der menschlichen Leistungsfähigkeit bei der Behandlung komplexer Krankheiten und der Bioproduktion.
Aus molekularer Sicht ist die Bindung eines Proteins an ein Zielmolekül im Wesentlichen eine dreidimensionale Strukturfrage:Die Aminosäurezusammensetzung, die räumliche Konformation und die intermolekularen Wechselwirkungen der Grenzfläche prägen gemeinsam die Affinität und Spezifität der Bindung.Daher kehren fast alle Methoden zur Bindemittelentwicklung letztendlich zur Kernvariablen „Struktur“ zurück und nutzen Strukturanalysen oder -vorhersagen, um den Molekülaufbau zu steuern.
In den letzten Jahren hat die Einführung von maschinellem Lernen dieses Paradigma grundlegend verändert. Dank Durchbrüchen bei Modellen zur Strukturvorhersage und -generierung löst sich die Forschung zunehmend von ihrer starken Abhängigkeit von experimentellen Strukturen und verlagert ihren Fokus von der „Strukturanalyse“ zur „Strukturgenerierung“. Dies ermöglicht die Entwicklung von Bindemitteln von Grund auf und reduziert F&E-Kosten und -Zeitaufwand erheblich.
Methodisch betrachtet weist die aktuelle KI-gestützte Binderentwicklung jedoch noch deutliche Unterschiede auf:Eine Art davon sind generative Methoden, wie sie beispielsweise durch RFDiffusion repräsentiert werden.Es ist auf umfangreiches Training angewiesen, um direkt Kandidatenstrukturen zu generieren, bietet aber nicht die Möglichkeit, sich während der Inferenzphase flexibel anzupassen;Eine andere Art ist die illusionäre Methode, die durch BindCraft repräsentiert wird.Gradientenoptimierung durch Bewertung von Strukturprädiktoren bietet zwar Flexibilität, jedoch fehlen generative Vorinformationen, was die Erforschung völlig neuer Strukturräume erschwert. Diese „Trennung von Generierung und Optimierung“ steht im Gegensatz zum einheitlichen Paradigma „vortrainierte Modelle + computergestützte Erweiterungen zur Inferenzzeit“, das in der Verarbeitung natürlicher Sprache und Bildverarbeitung bereits etabliert ist.
In diesem ZusammenhangEin gemeinsames Forschungsteam von NVIDIA, der Universität Oxford, dem Quebec Artificial Intelligence Institute und anderen Institutionen hat das Proteina-Complexa-Framework (im Folgenden Complexa genannt) vorgeschlagen.Dieser Ansatz zielt darauf ab, die Lücke zwischen generativen und illusionären Methoden zu schließen, indem er das grundlegende generative Modell und die Mechanismen zur Optimierung der Inferenzzeit in einem einzigen System vereint. Er basiert auf Teddymer-Vortraining.Complexa ermöglicht die Entwicklung hochmoderner, neuartiger Bindemittel, ohne dass zusätzliche Schritte zur Neugestaltung der Sequenz erforderlich sind.Durch die Anpassung der Testzeit-Skalierungstechnik aus dem Diffusionsmodell an diesen Rahmen werden Generierung und Optimierung direkt vereinheitlicht, wodurch traditionelle illusionsbasierte Methoden hinsichtlich der Leistung übertroffen werden.
Die zugehörigen Forschungsergebnisse mit dem Titel „Scaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute“ wurden für die ICLR 2026 angenommen.
Forschungshighlights:
* Diese Studie schlägt Complexa vor, das La-Proteina auf das Binder-Design erweitert, Teddymer nutzt und eine effiziente Inferenzzeitoptimierung erreicht, die durch generative Priors beschleunigt wird.
* Erzielte Spitzenwerte bei Computersimulationen in Benchmarks für Protein- und Kleinmolekül-Target- und Enzymdesign, ohne dass eine Sequenzneugestaltung erforderlich war.
Papieradresse:
https://openreview.net/forum?id=qmCpJtFZra
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „Complexa“, um das vollständige PDF zu erhalten.
Datensätze: Von der „Einzelzell-Anreicherung“ zur „Komplexen Rekonstruktion“
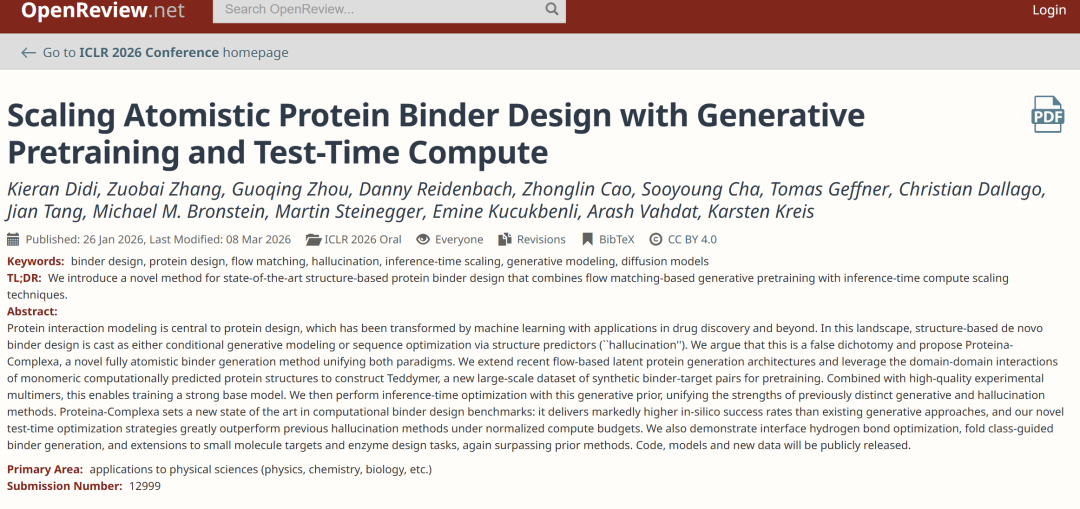
Eine grundlegende Einschränkung von Bindergenerierungsmodellen liegt in den Daten. Idealerweise benötigt das Modell eine große Menge an Daten zu „Bindemittel-Zielprotein-Komplexen“ für das Training. In der Realität stammen diese Daten jedoch hauptsächlich aus experimentell analysierten Proteindatenbanken (PDBs), die in ihrem Umfang begrenzt sind und in denen qualitativ hochwertige Proben noch seltener vorkommen. Die größere AlphaFold-Datenbank (AFDB) bietet zwar eine enorme Menge an Proteinstrukturen, doch handelt es sich dabei fast ausschließlich um monomere Strukturen, denen Informationen über komplexe Proteine fehlen.Diese strukturelle Diskrepanz zwischen „reichlich vorhandenen Einzelkomponenten und wenigen komplexen Komponenten“ schränkt die Fähigkeit des Modells, in großem Umfang trainiert zu werden, unmittelbar ein.
Der entscheidende Durchbruch dieser Studie liegt in einem neuen Verständnis der inneren Struktur der AFDB. Die meisten Proteine in der AFDB sind Multidomänenproteine, die mithilfe des Domänensegmentierungstools TED detailliert annotiert werden. Weiterführende Analysen zeigten, dass die Wechselwirkungen zwischen verschiedenen Domänen innerhalb desselben Proteins statistisch ähnliche Merkmale aufweisen wie jene in mehrsträngigen Komplexen. Diese Beobachtung führt zu einem wichtigen Paradigmenwechsel:Monomerstrukturen sind an sich keine „nutzlosen Daten“, sondern können als potenzielle Datenquelle für komplexe Strukturen neu interpretiert werden.
Auf dieser Grundlage schlägt die Studie eine Methode zur „künstlichen Multimerkonstruktion“ vor.Indem man Multidomänenproteine in unabhängige Domänen aufteilt und diese als unterschiedliche Ketten behandelt, kann innerhalb des Monomers eine komplexartige Struktur aufgebaut werden.Der spezifische Prozess beginnt mit AFDB50, wobei Proteine mit TED-Annotationen gescreent, anschließend in „Pseudo-Multimere“ aufgeteilt, Dimere extrahiert und anhand ihrer räumlichen Nähe gefiltert werden, wobei Proben mit vollständigen Annotationen beibehalten werden. Nach dem Clustering zur Entfernung von Redundanzen werden schließlich etwa 3,5 Millionen Dimercluster erhalten. Dieser Datensatz trägt den Namen Teddymer und erweitert nicht einfach nur den Datenumfang, sondern wandelt den „Monomervorteil“ durch strukturelle Reorganisation in ein „Komplexangebot“ um.
Während des Trainingsprozesses, wie in der folgenden Abbildung dargestellt, stützte sich die Studie nicht auf eine einzige Datenquelle, sondern integrierte AFDB-Monomerdaten, Teddymer-Konstruktionsdaten, PDB-Experimentalkomplexdaten und PLINDER-Protein-Ligand-Daten. Dadurch konnte das Modell eine einheitliche Darstellung zwischen Monomerstruktur, Komplexstruktur und Wechselwirkungen kleiner Moleküle herstellen und somit sowohl generative als auch generalisierende Fähigkeiten berücksichtigen.
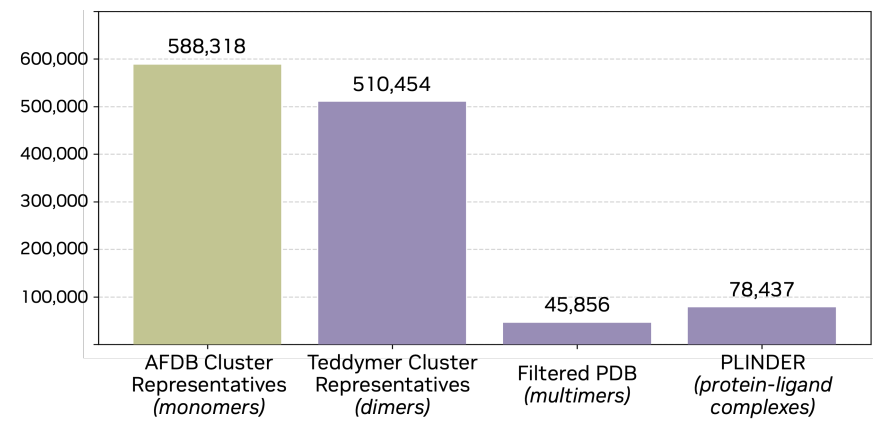
Complexa: Ein vollständig atomares Gerüst zur Generierung von Proteinbindungsmitteln
Im Hinblick auf das Modelldesign besteht die Kernänderung bei Complexa nicht nur in einer „stärkeren Generierungsfähigkeit“, sondern vielmehr in einer Verlagerung des generativen Ziels von „vollständigen Proteinstrukturen“ hin zu „Bindemitteln an spezifischen Grenzflächen“. Aufbauend auf La-Proteína liegt sein Vorteil in seiner Fähigkeit, auf der gesamten atomaren Ebene zu generieren.Gleichzeitig vermeidet es dank der effizienten Transformer-Architektur Module mit hohem Rechenaufwand in traditionellen Strukturmodellen, was ihm eine gute Skalierbarkeit in groß angelegten Stichprobenszenarien verleiht.
Darauf aufbauend stellt die Studie einen Generierungsmechanismus vor, der auf Zielpunkten und Hotspots an der Schnittstelle basiert. Dieser Mechanismus verhindert, dass das Modell vollständige Komplexe generiert, sondern nur den Binderanteil, und nutzt während des Generierungsprozesses explizit Zielinformationen. Konkret:Das Flow-Matching-Modell ist für die Generierung von Strukturen unter bedingten Einschränkungen zuständig, während der Autoencoder nur zum Codieren und Decodieren von Monomerbindern verwendet wird, wodurch die Modellkomplexität reduziert und gleichzeitig die Ausdruckskraft erhalten bleibt.
Um dem Modell ein effektives Verständnis der Zielinformationen zu ermöglichen, wurde ein systematisches Design für die Eingaberepräsentation implementiert. Proteinziele wurden mithilfe der Atom37-Methode kodiert, wobei 3D-Koordinaten auf Restebene, Aminosäuretypen und Hotspot-Informationen einheitlich in das Modell eingespeist wurden. Hotspots kennzeichnen potenzielle Bindungsregionen. Während des Trainings wurden diese Hotspots aus realen Schnittstellen extrahiert, während sie während der Inferenz als Priors verwendet oder durch Vorverarbeitung gewonnen wurden. Für niedermolekulare Ziele kodierte das Modell Typ, Ladung und räumliche Koordinaten auf atomarer Ebene. Diese wurden zusammen mit der Repräsentation des Bindungsagenten in den Transformer für die gemeinsame Modellierung eingegeben.
Bezüglich der AusbildungszieleEine wesentliche Verbesserung ist die Einführung von zufälligem globalem Translationsrauschen in die Binderkoordinaten, wodurch das Modell gezwungen wird, die räumliche Lokalisierungsfähigkeit der Moleküle zu erlernen.Dies ist für die Monomergenerierung nicht relevant, stellt aber eine Schlüsselfunktion dar, die die Generierungsqualität für Aufgaben bestimmt, die eine präzise Platzierung des Binders an der Zielschnittstelle erfordern. Der gesamte Trainingsprozess folgt einer phasenweisen Strategie und schreitet schrittweise von der Monomermodellierung über die Generierung allgemeiner Strukturen bis hin zum binderspezifischen Training fort. Gleichzeitig wird Overfitting durch LoRA kontrolliert, und der Autoencoder auf Monomerebene wird durchgehend wiederverwendet, um die Architektur einfach zu halten.
In der InferenzphaseComplexa führt außerdem einen „Testzeit-Berechnungserweiterungsmechanismus“ ein, der den Generierungsprozess mit der Suchoptimierung kombiniert.Durch die Erhöhung der Stichprobenanzahl, die Einführung von Bündelsuche oder Monte-Carlo-Baumsuche kann das Modell die Qualität seiner Generierung bei einem größeren Rechenbudget kontinuierlich verbessern. Dieser Ansatz ermöglicht es dem Modell, seine Fähigkeiten während der Inferenz dynamisch zu erweitern, anstatt ausschließlich auf die Trainingsphase beschränkt zu sein.
Höhere Erfolgsquote, schnellere Bearbeitung und bessere Skalierbarkeit
Um die Leistungsfähigkeit des Modells zu validieren, wurde in dieser Studie eine Reihe von Experimenten mit unterschiedlichem Schwierigkeitsgrad durchgeführt. Die zentrale Frage lautet: Übertrifft Complexa nicht nur die grundlegenden Anforderungen, sondern verbessert sich seine Leistung auch kontinuierlich mit zunehmenden Rechenressourcen?
Im Hinblick auf die grundlegenden Erzeugungskapazitäten,Egal ob es sich um die Zielsetzung von Proteinen oder kleinen Molekülen handelt, Complexa übertrifft bestehende Methoden deutlich und zeichnet sich durch höhere Erfolgsraten und schnellere Probenahmegeschwindigkeiten aus.Gleichzeitig wird die Neuartigkeit der generierten Strukturen deutlich verbessert. Noch wichtiger ist, dass das Modell direkt hochwertige Sequenzen ausgeben kann, ohne auf Tools wie ProteinMPNN für das sekundäre Design angewiesen zu sein, wodurch der Gesamtprozess vereinfacht wird.
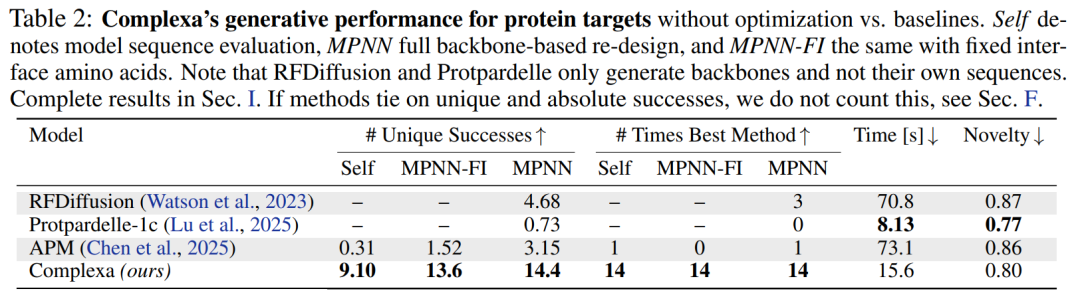
Im Hinblick auf die strukturelle Kontrollierbarkeit,Die Studie führt bedingte Bezeichnungen ein, die es dem Modell ermöglichen, den Typ der generierten Struktur explizit zu steuern.Die Wahl zwischen α-Helices und β-Faltungen kann beispielsweise das Problem der einheitlichen Struktur bisheriger generativer Modelle wirksam beheben und die strukturelle Vielfalt deutlich verbessern.
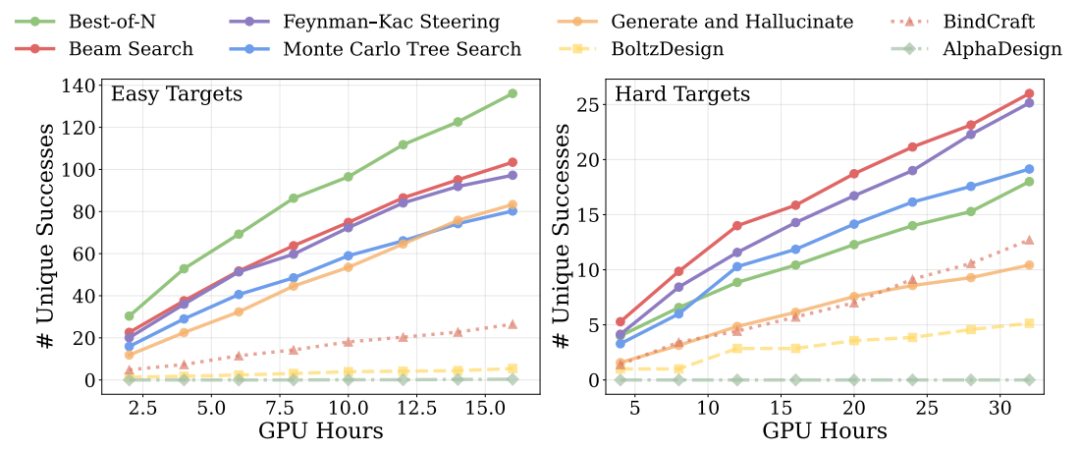
Die Ergebnisse von Computerexperimenten zur Erweiterung der Inferenzphase zeigen, dass bei einfachen Aufgaben bereits eine Erhöhung der Anzahl der Stichproben ausreicht, um alle Basismethoden zu übertreffen, während bei komplexen Aufgaben die Einführung fortgeschrittenerer Suchstrategien (wie Bündelsuche und Monte-Carlo-Baumsuche) den Vorteil noch weiter ausbaut.Dies deutet darauf hin, dass sich die Modellleistung mit zunehmendem Rechenbudget weiter verbessern lässt.Wie in der Abbildung unten gezeigt
Hinsichtlich der physikalischen Plausibilität sind weitere Untersuchungen erforderlich, um die Wasserstoffbrückenbindungen an den Grenzflächen und die damit verbundenen Energieindizes zu optimieren.Es zeigte sich, dass das Modell nicht nur strukturell einwandfreie Bindemittel erzeugen, sondern diese auch auf der Ebene der feinkörnigen Wechselwirkungen optimieren konnte.Dies verbessert die Stabilität der Bindung.
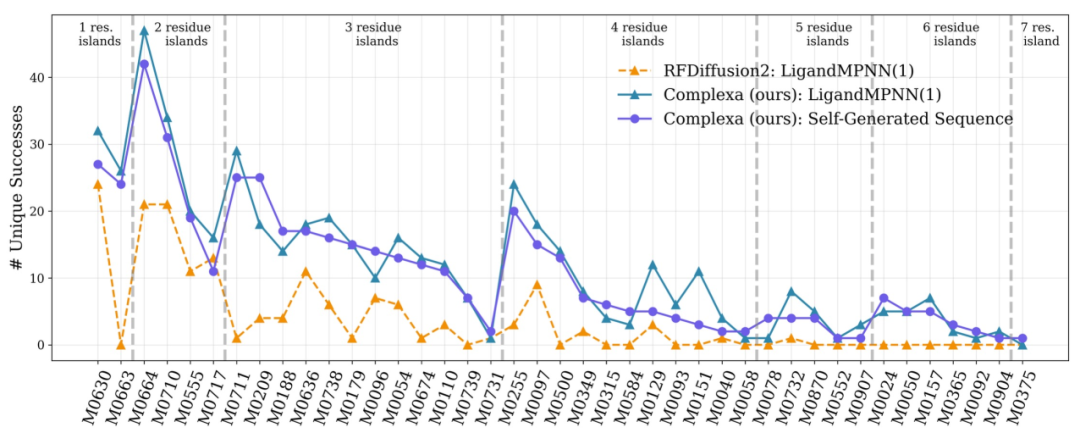
Bei anspruchsvolleren Multi-Chain-Zielaufgaben können die bestehenden Methoden unter begrenzten Rechenressourcen keine effizienten Lösungen erzielen.Complexa generierte nach der Erweiterung der Rechenressourcen erfolgreich qualitativ hochwertige Kandidaten und demonstrierte damit seine Skalierbarkeit bei komplexen Problemen.Die Ergebnisse der Tests zu verschiedenen Aufgaben, wie beispielsweise dem Enzymdesign, sind in der folgenden Abbildung dargestellt. Das Framework zeichnet sich durch eine gute Generalisierungsfähigkeit aus und lässt sich vom Binderdesign auf ein breiteres Spektrum von Problemen im Bereich des Protein-Engineerings ausweiten.
Paradigmenwechsel im KI-Protein-Design
Die Entwicklung KI-gestützter Proteinbindungsmittel hat in den letzten Jahren rasant Fortschritte gemacht und ist von der Theorie in die Praxis übergegangen. Nobelpreisträger David Baker und sein Team zählen weiterhin zu den führenden Experten auf diesem Gebiet. Im Jahr 2025 veröffentlichte ihr Team mehrere Studien in der Fachzeitschrift Science.Das System bestätigte die Machbarkeit der Entwicklung hochspezifischer pMHC-Binder auf Basis von RFDiffusion.Die verwandten Arbeiten befassten sich mit 11 Krankheitskategorien, erzeugten erfolgreich Bindungsproteine, die T-Zellen dazu bringen können, Tumore zu erkennen, und verifizierten die Genauigkeit des Designs auf atomarer Ebene mittels Kryo-Elektronenmikroskopie, was den Beginn der Verifizierbarkeit für das KI-Design markiert.
Das MIT-Team verfolgte derweil einen stärker integrierten Ansatz im BoltzGen-Modell, indem es die Strukturvorhersage und die Aggregatgenerierung in einem einzigen All-Atom-Modell vereinte und kontinuierliche geometrische Darstellungen anstelle der traditionellen diskreten Modellierung verwendete.In Experimenten mit 26 Zielmolekülen erreichte 66% als Binder eine nanomolare Affinität.Es weist auch bei Zielen außerhalb des regulären Vertriebsnetzes eine hohe Erfolgsquote auf und beweist damit eine gewisse Generalisierungsfähigkeit.
Die Branche konzentriert sich zunehmend auf die technische Umsetzung dieser Fähigkeiten. Anfang 2026 schlossen Bayer und Cradle eine dreijährige Kooperation, um ihre KI-basierte Protein-Engineering-Plattform in den Antikörperentwicklungsprozess zu integrieren. Diese Plattform wurde bereits in über 50 Projekten eingesetzt, wodurch Entwicklungszyklen deutlich verkürzt und ein iterativer, geschlossener Prozess aus Design, Test und Lernen unterstützt werden konnten. Dies verdeutlicht, dass sich KI von einem Hilfsmittel zu einer grundlegenden Fähigkeit im F&E-Prozess entwickelt.
Insgesamt verlagert sich der Fokus im Proteindesign von der Leistungsfähigkeit einzelner Modelle hin zu Systemeffizienz und Skalierbarkeit. Die akademische Forschung erweitert kontinuierlich die Grenzen der Modellleistung, während die Industrie einen stabilen und wiederverwendbaren F&E-Prozess vorantreibt. KI-gestütztes Proteindesign tritt somit in eine praxisorientiertere Phase ein: Entscheidend ist nicht mehr, ob ein Protein designt werden kann, sondern ob ein kontinuierliches und effizientes Design möglich ist.
Referenzlinks:
1.https://news.bioon.com/article/00bf92186439.html
2.https://mp.weixin.qq.com/s/1zKXUQtXgCJ7GA1_OUEShg
3.https://www.bayer.com/en/us/news-stories/ai-enabled-antibody-discovery-and-optimization