Command Palette
Search for a command to run...
KI-gestützte Quantenverfeinerung: Die Carnegie Mellon University Und Andere Stellen AQuaRef Vor, Das Erste System, Das Quantenmechanische Randbedingungen Nutzt, Um Das Gesamtatommodell Eines Proteins Zu verfeinern.
Um die molekularen Mechanismen von Lebensprozessen zu verstehen, müssen wir zunächst die dreidimensionale Struktur biologischer Makromoleküle betrachten.Die Bestimmung atomarer Strukturen ist eine Kernaufgabe der Strukturbiologie und eine wichtige Grundlage für das Verständnis der Proteinfunktion, die Aufdeckung genetischer Regulationsmechanismen und die Entwicklung zielgerichteter Medikamente.Ob es sich um proteinkatalysierte Reaktionen, die Übertragung genetischer Informationen durch Nukleinsäuren oder die Erkennung von Antigenen durch Antikörper handelt – all diese wichtigen biologischen Prozesse benötigen präzise Strukturmodelle zur Erklärung.
Aktuell sind Kryo-Elektronenmikroskopie und Röntgenkristallographie die wichtigsten experimentellen Techniken zur Aufklärung der Strukturen biologischer Makromoleküle, und es wurde eine große Menge hochauflösender Strukturdaten gesammelt. In den letzten Jahren haben auch computergestützte Vorhersagemethoden, wie beispielsweise AlphaFold und RoseTTAFold, bedeutende Fortschritte erzielt und bieten effiziente Werkzeuge für die Strukturmodellierung. Dennoch spielt die experimentelle Analyse weiterhin eine unersetzliche Rolle bei der Entdeckung unbekannter Strukturtypen und der Aufklärung komplexer Wechselwirkungen.Im experimentellen Strukturaufklärungsprozess ist die Verfeinerung des Atommodells ein entscheidender Schritt, der nahe am Ende liegt. Ziel ist es, ein Molekülmodell zu erstellen, das den Gesetzen der Stereochemie entspricht und die experimentellen Daten bestmöglich wiedergibt.Gängige Verfeinerungsprogramme wie CCP4 und Phenix stützen sich hauptsächlich auf stereochemische Beschränkungen in Standarddatenbanken, um sinnvolle Bindungslängen und -winkel beizubehalten und interatomare Konflikte zu reduzieren.
Solche Restriktionssysteme weisen jedoch noch erhebliche Einschränkungen auf. Sie zielen primär auf kovalente Strukturen ab und vernachlässigen eine systematische Beschreibung wichtiger nicht-kovalenter Wechselwirkungen wie Wasserstoffbrückenbindungen und π-Wechselwirkungen. Bei niedriger Auflösung kann dies zu Modellen führen, die vom tatsächlichen chemischen Zustand abweichen. Treten neue Liganden oder einzigartige Verbindungen in der Struktur auf, ist eine manuelle Parameterdefinition zur Verfeinerung erforderlich. Darüber hinaus können plausible geometrische Abweichungen, die durch die lokale chemische Umgebung bedingt sind, vom Restriktionssystem fälschlicherweise als Anomalien interpretiert und zwangsweise korrigiert werden. Theoretisch …Die Quantenmechanik kann intermolekulare Wechselwirkungen genauer beschreiben, aber biologische Makromoleküle enthalten typischerweise Tausende oder sogar Zehntausende von Atomen, was das vollständige Quantencomputing extrem teuer macht.Daher beschränken sich die meisten bestehenden Studien auf lokale Bereiche wie Ligandenbindungsstellen.
Um dieses Problem anzugehen, wurde ein gemeinsames Forschungsteam der Carnegie Mellon University, der Universität Breslau in Polen und der University of Florida sowie anderer Universitäten gegründet.Es wird eine KI-gestützte Quantenverfeinerungsmethode, AQuaRef, vorgeschlagen.Diese Methode basiert auf dem maschinellen Lernen von Atompotentialfunktionen mit AIMNet2 und wurde speziell für die Verfeinerung angepasst. Sie erreicht annähernd die Recheneffizienz klassischer Kraftfelder und kann die Ergebnisse quantenmechanischer Berechnungen besser approximieren. Damit eröffnet sie einen neuen technischen Weg für die atomare Quantenverfeinerung biologischer Makromoleküle.
Die zugehörigen Forschungsergebnisse mit dem Titel „AQuaRef: maschinelles Lernen beschleunigte Quantenverfeinerung von Proteinstrukturen“ wurden in Nature Communications veröffentlicht.
Forschungshighlights:
* AQuaRef, basierend auf der AIMNet2-Maschinenlernpotentialfunktion, erreicht erstmals eine Quantenverfeinerung eines vollständigen Proteinatommodells.
* In Tests von 61 Modellen für die Röntgen- und Kryo-Elektronenmikroskopie mit niedriger Auflösung schnitt AQuaRef bei 57 Modellen besser ab.
* Im Falle kurzer Wasserstoffbrückenbindungen in den Proteinen DJ-1 und YajL kann AQuaRef Protonenpositionen bestimmen, die mit experimentellen Befunden übereinstimmen, ohne dass ein menschliches Eingreifen erforderlich ist.

Papieradresse:https://www.nature.com/articles/s41467-025-64313-1
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „AQuaRef“, um das vollständige PDF zu erhalten.
Ein Datensatz von 1 Million Proben zum Trainieren der Potenzialfunktion im maschinellen Lernen von Peptiden.
Ziel dieser Studie ist es, mithilfe von maschinellem Lernen ein parametrisiertes Modell der Potentialfunktion für ein Peptidsystem zu erstellen.Daher muss das Datensatzdesign systematisch drei Dimensionen abdecken: chemische Zusammensetzung, Konformationsraum und intermolekulare Wechselwirkungen.
Aus chemischer Sicht,Die Forscher erstellten eine kleine Peptiddatenbank in Form von SMILES-Strings, die 20 Standard-Aminosäuren, 11 Protonierungszustände, 3 N-terminale Modifikationen und 4 C-terminale Modifikationen abdeckt.Aufbauend auf dieser Grundlage wurden alle Einzel- und Dipeptide aufgelistet und eine Untergruppe von Tri- und Tetrapeptiden zufällig ausgewählt. Zusätzlich wurden über Disulfidbrücken verknüpfte Peptide und ihre selenisierten Analoga generiert. Um den gesamten Konformationsraum abzudecken, nutzten die Forscher die Software OpenEye Omega für eine intensive Torsionswinkelmessung ohne Einschränkungen hinsichtlich chiraler Zentren. Dadurch ist das Modell auf D-, L- und gemischt stereochemische Peptidsysteme anwendbar.
Es wurden Komplexe aus 2–4 Peptiden konstruiert und deren räumliche Orientierung zufällig variiert, um intermolekulare Wechselwirkungen zu simulieren. Der gesamte Datengenerierungsprozess basierte nicht auf natürlichen Sequenzen oder experimentellen Strukturen, um potenzielle Datenlecks zu vermeiden. Zur Kontrolle des Rechenaufwands wurde die Gesamtzahl der Atome (einschließlich Wasserstoff) in allen Peptiden und ihren Komplexen auf 120 begrenzt.
Nach Erhalt der AusgangskonformationForscher nutzten zunächst GFN-FF-Kraftfelder, um Molekulardynamiksimulationen durchzuführen und so Nichtgleichgewichtsstrukturen zu untersuchen.Es erhält die Gesamtkonfiguration nahe am ursprünglichen Eingangszustand, indem es sie mit kartesischen Koordinaten einschränkt, während es gleichzeitig den Torsionswinkel und die intermolekularen Freiheitsgrade freigibt.
Anschließend wurde eine aktive Lernstrategie mittels Query-by-Committee eingeführt: Zunächst wurden 500.000 Ausgangsproben zufällig ausgewählt, um ein Ensemble-System aus vier Modellen zu trainieren. Danach wurden vier Iterationen durchgeführt. In jeder Iteration wurden Proben basierend auf der Unsicherheit der Modellvorhersagen von Energie und Atomkräften ausgewählt. Diese Strukturen mit hoher Unsicherheit wurden nach DFT-Berechnungen dem Trainingsdatensatz hinzugefügt. In der letzten Iteration wurde zusätzlich eine unsicherheitsgeleitete Optimierung eingeführt, die Grenzstrukturen mit hoher Vorhersageunsicherheit, aber niedriger Energie priorisierte. Durch diesen Prozess wurde schließlich ein Trainingsdatensatz von etwa 1 Million Proben mit durchschnittlich etwa 42 Atomen erhalten.
Zusätzlich zu theoretisch generierten Daten nutzten die Forscher experimentelle Strukturen aus den Datenbanken RCSB und EMDB zur Validierung ihrer Modelle. Zu den Auswahlkriterien gehörten: Modelle mit einer einzigen Konformation, die ausschließlich Proteine enthielten, 1.000–10.000 Nicht-Wasserstoffatome, eine Auflösung von 2,5–4 Å, ein MolProbity-Konfliktwert von unter 50 sowie Abweichungen der Bindungslängen und -winkel von maximal dem Vierfachen der Standardwerte.
AQuaRef: KI-gestützte Quantenverfeinerungsansätze für makromolekulare Systeme
AQuaRef führt zunächst eine Integritätsprüfung des eingegebenen Atommodells durch. Fehlende Atome in der Struktur werden vom Programm automatisch ergänzt. Dieser Prozess kann jedoch mitunter neue sterische Konflikte verursachen, insbesondere wenn das ursprüngliche Modell keine Wasserstoffatome enthält. Handelt es sich bei den fehlenden Atomen um kritische Strukturen wie Hauptkettenatome, kann die Quantenverfeinerung nicht fortgesetzt werden. Werden signifikante sterische Konflikte oder gravierende geometrische Anomalien festgestellt, wird zunächst eine schnelle geometrische Regularisierung mithilfe standardmäßiger stereochemischer Beschränkungen durchgeführt, um das Problem mit minimalen Anpassungen der Atompositionen zu beheben.
Bei der Verfeinerung kristallographischer Daten müssen auch die Symmetrie der Einheitszelle und periodische Wechselwirkungen berücksichtigt werden.Das Programm erweitert das Modell mithilfe des Symmetrieoperators der Raumgruppe zu einer Superzelle und beschränkt diese anschließend, wobei nur die symmetrischen Kopien beibehalten werden, deren Abstand zum Hauptkopieatom innerhalb eines festgelegten Bereichs liegt. Dieser Prozess ist in der Regel bei Kryo-Elektronenmikroskopie-Strukturen nicht erforderlich.
Nach Abschluss der atomaren Ergänzung und Modellerweiterung durchläuft das System den Standard-Verfeinerungsprozess des Q|R-Softwarepakets. Die Kernarchitektur von AQuaRef entspricht im Wesentlichen der des Basismodells AIMNet2, jedoch wurden für die Strukturverfeinerung einige wichtige Anpassungen vorgenommen.
Erstens berechnet das Modell nicht explizit langreichweitige Coulomb- und dispersive Wechselwirkungen, sondern wird direkt darauf trainiert, die gesamte DFT-D4-Energie zu reproduzieren.Dies liegt daran, dass die Coulomb-Wechselwirkung im CPCM-Modell mit implizitem Lösungsmittel allein anhand der Atomladung nur schwer genau abgeschätzt werden kann und die Fernwechselwirkung durch das polarisierbare Kontinuum stark abgeschirmt wird. Zudem tragen die Ferndispersionskräfte mit einem Abschneideradius von mehr als 5 Å nur sehr geringfügig zu den wichtigsten Atomkräften im Verfeinerungsprozess bei, sodass sie ohne Beeinträchtigung der Genauigkeit vernachlässigt werden können.
Zweitens führt das Modell einen expliziten exponentiellen Abstoßungsterm mit kurzer Reichweite aus GFN1-XTB ein, was zu einer besseren Stabilität bei Strukturen mit räumlichen sterischen Hinderungskonflikten führt.Das Modelltraining erfolgte unter Verwendung der Energie, der Atomkraft und der partiellen Hershfield-Ladung der Atome, berechnet mit der B97M-D4/def2-QZVP-Methode. Das Training begann mit zufälliger Gewichtsinitalisierung, einer Batchgröße von 256 und insgesamt 1,5 Millionen Trainingsschritten. Alle anderen Hyperparameter wurden von den ursprünglichen AIMNet2-Einstellungen übernommen.
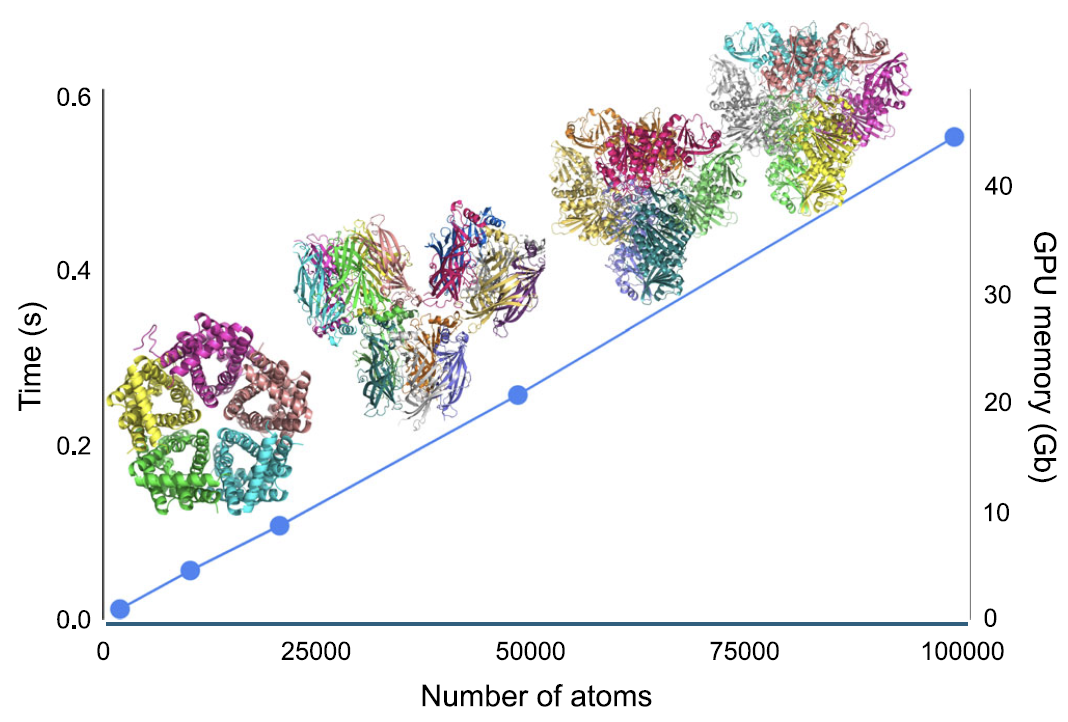
Hinsichtlich der Recheneffizienz, wie in der folgenden Abbildung dargestellt...Im AIMNet2-Framework steigen sowohl die Rechenzeit für Energie und Atomkräfte als auch die maximale GPU-Speicherauslastung linear (O(N)) mit der Anzahl der Atome im System an.Für ein Proteinsystem mit etwa 100.000 Atomen benötigen Einzelpunkt-Energie- und Kraftberechnungen nur etwa 0,5 Sekunden; auf einer einzelnen NVIDIA H100 GPU mit 80 GB Videospeicher können Modelle mit bis zu etwa 180.000 Atomen verarbeitet werden.
Die lokale Struktur von AQuaRef wurde anhand von 41 Kryo-Elektronenmikroskopie- und 20 Röntgenmodellanalysen auf 2 Å optimiert.
Um die Leistung von AQuaRef zu bewerten,Die Forscher erstellten einen Testdatensatz, der 41 Kryo-Elektronenmikroskopie-Modelle, 20 niedrigauflösende und 10 ultrahochauflösende Röntgenmodelle umfasste.Alle 61 niedrigauflösenden Modelle verfügen über entsprechende hochauflösende homologe Referenzstrukturen. Während des Verfeinerungsprozesses wurden drei Arten von Randbedingungen zum Vergleich festgelegt: AIMNet2-Quantenbeschränkungen (d. h. AQuaRef), standardmäßige geometrische Beschränkungen sowie zusätzliche Beschränkungen wie Wasserstoffbrückenbindungen und Sekundärstrukturen.
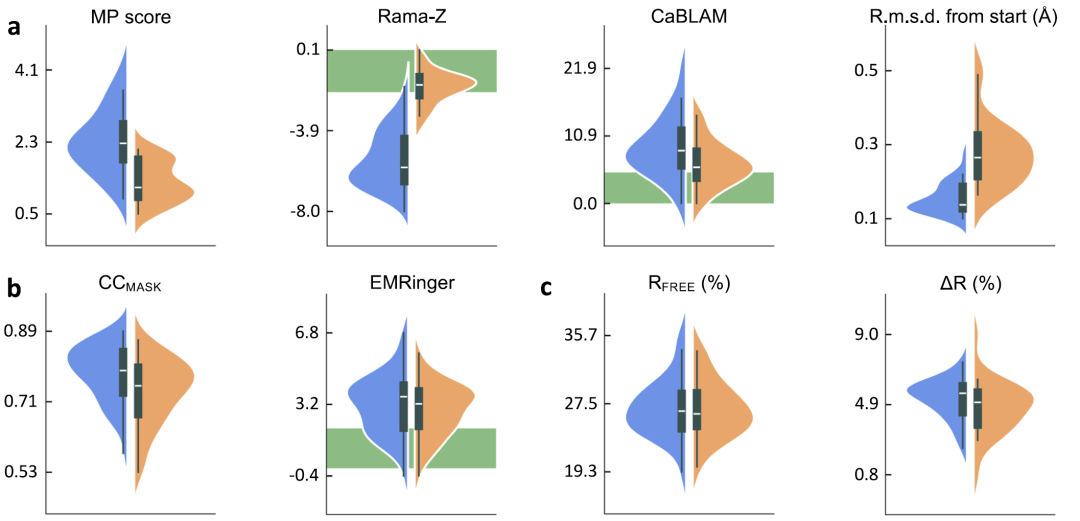
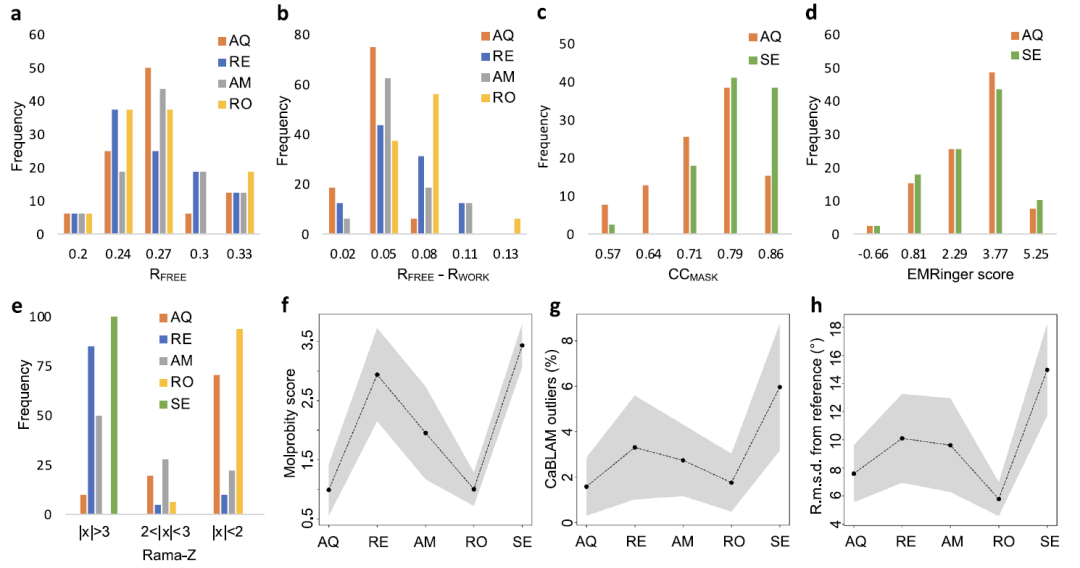
Die Ergebnisse sind in der folgenden Abbildung dargestellt.Das quantenverfeinerte Niedrigauflösungsmodell übertrifft herkömmliche Constraint-Methoden in geometrischen Metriken wie dem MolProbity-Score und dem Ramachandran-Plot-Z-Score deutlich.Die Anpassung des Modells an die experimentellen Daten blieb unterdessen weitgehend konstant. Bei Röntgenstrukturen wurde die Überanpassung leicht reduziert (die Differenz zwischen R<sub>work</sub> und R<sub>free</sub> war geringer); bei Kryo-Elektronenmikroskopie-Strukturen sank CC<sub>mask</sub> leicht, während der EMRinger-Wert im Wesentlichen unverändert blieb. Zusammen mit der insgesamt verbesserten geometrischen Qualität deuten diese Ergebnisse darauf hin, dass die Überanpassung des Modells verringert worden sein könnte.
Obwohl zusätzliche geometrische Randbedingungen die Modellqualität verbessern können, liefert AQuaRef weiterhin plausiblere Geometrien und liegt näher am hochauflösenden Referenzmodell. In einigen Fällen kann die lokale Abweichung zwischen den Standardrandbedingungen und der quantenverfeinerten Struktur bis zu 2 Å betragen.
Die Studie verglich AQuaRef außerdem mit mehreren gängigen Raffinationsmethoden. Die Ergebnisse sind in der folgenden Abbildung dargestellt. AMBER, Rosetta und REFMAC5 wurden für Röntgendaten ausgewählt, während Servalcat für Kryo-Elektronenmikroskopie-Daten verwendet wurde. InsgesamtAQuaRef bietet eine etwas bessere Leistung als Rfree und weist den geringsten Grad an Überanpassung auf.Im Vergleich zu Servalcat weisen beide vergleichbare EMRinger-Werte auf, Servalcat hat jedoch einen etwas höheren CCmask-Wert.
In Bezug auf die geometrische Qualität,AQuaRef verhält sich ähnlich wie Rosetta, ist aber REFMAC5 und Servalcat deutlich überlegen.Rosetta weist eine etwas bessere Gesamtübereinstimmung mit dem Referenzmodell auf, was mit dem größeren Konvergenzradius aufgrund der gradientenfreien Optimierungsstrategie zusammenhängen könnte. Darüber hinaus können AQuaRef und Rosetta plausible Wasserstoffbrückenbindungen generieren, gefolgt von AMBER, während REFMAC5 und Servalcat diese Details im Grunde nicht präzise wiedergeben können.
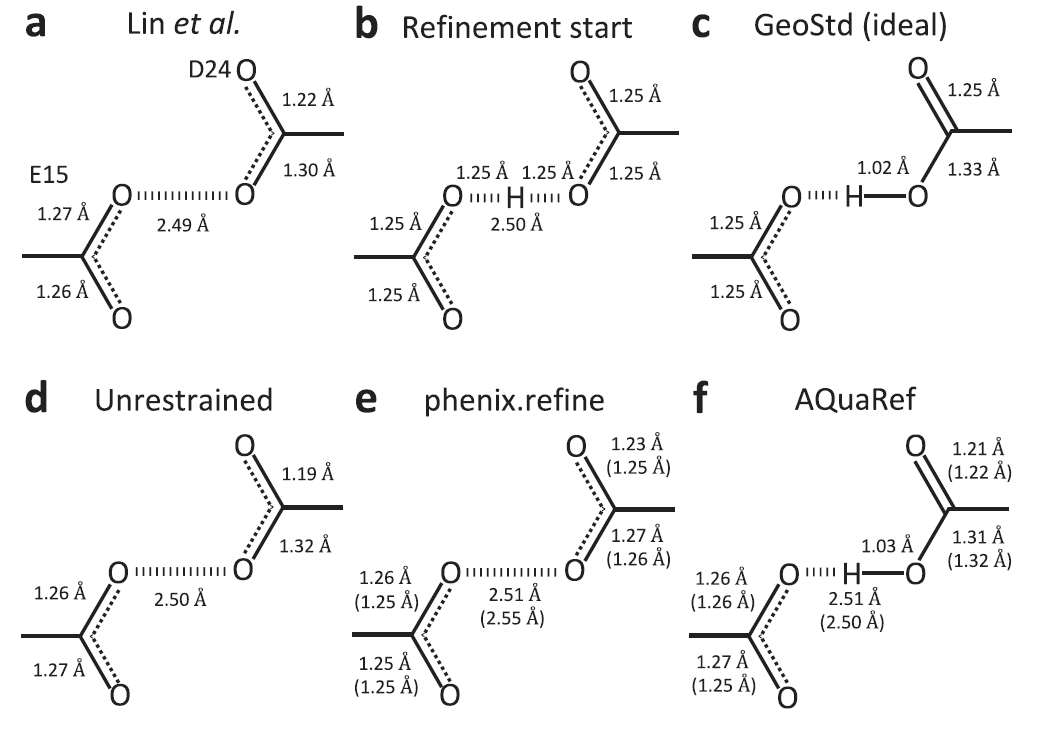
In Tests an Systemen mit kurzen Wasserstoffbrückenbindungen nutzten Forscher das Parkinson-assoziierte Protein DJ-1 und sein Homolog YajL als Beispiele, um die Fähigkeit von AQuaRef zur Behandlung protonierter Zustände zu untersuchen. Traditionelle Verfeinerungsmethoden, die durch die Stereochemie der Datenbank eingeschränkt sind, führen häufig zu Abweichungen der Bindungslängen von ihren tatsächlichen Werten.Bei Verwendung der symmetrischen diprotonierten Struktur als Ausgangsmodell für die AQuaRef-Verfeinerung stimmen die resultierenden Protonenpositionen und die Bindungsgeometrie mit den Ergebnissen der unbeschränkten Verfeinerung überein.Durch die Hinzunahme konventioneller Randbedingungen werden die Bindungslängen an die deprotonierten Standardwerte in der Datenbank angenähert. Selbst wenn experimentelle Daten auf eine Auflösung von 2 Å reduziert werden, wodurch die atomaren Details deutlich verringert werden, kann AQuaRef eine Struktur rekonstruieren, die nahezu identisch mit den ursprünglichen 1,15 Å-Daten ist, während die Verfeinerung mit konventionellen Randbedingungen weiter von der tatsächlichen Konfiguration abweicht. AQuaRef positioniert das Proton am Oδ2-Atom des D24-Restes in DJ-1, ein Ergebnis, das sowohl durch Energieberechnungen als auch durch differentielle Elektronendichtekarten gestützt wird.
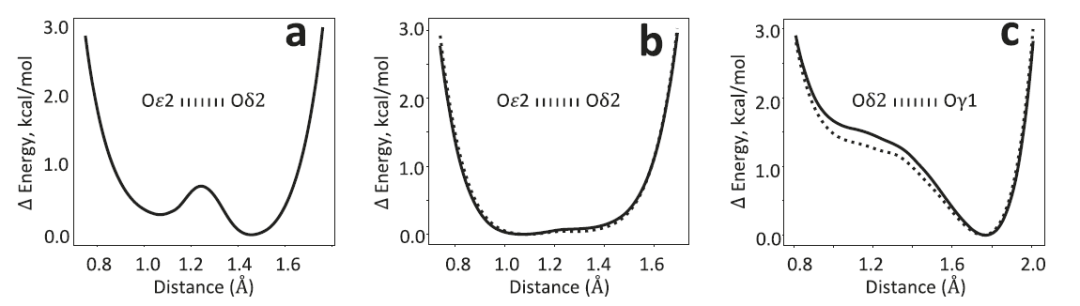
Im YajL-Protein stimmen die AQuaRef-Verfeinerungsergebnisse für die beiden kurzen Wasserstoffbrückenbindungen zwischen E14 und D23 mit der unbeschränkten Verfeinerung überein. Dies deutet darauf hin, dass das Proton sowohl von D23 als auch von E14 geteilt wird und typische Eigenschaften einer Wasserstoffbrückenbindung mit niedriger Barriere aufweist. Dies unterscheidet sich von DJ-1, wo das Proton hauptsächlich an einem einzelnen Sauerstoffatom lokalisiert ist. Die von AIMNet2 berechnete Energieverteilung zeigt eine relativ flache Potentialenergiefläche, was bedeutet, dass die Protonenposition unter Berücksichtigung der experimentellen Daten frei angepasst werden kann. Gleichzeitig zeigt die differentielle Elektronendichtedarstellung deutlich höhere Peaks als 3σ in der Nähe des Wasserstoffatoms, was diese strukturelle Interpretation weiter untermauert.
Bahnbrechende Fortschritte in der Zusammenarbeit zwischen Industrie, Wissenschaft und Forschung auf dem Gebiet der Quantenverfeinerung von Proteinen
In den zukunftsweisenden Bereichen der Quantenverfeinerung von Proteinen, der Konstruktion von Potenzialfunktionen mittels maschinellen Lernens und der Optimierung atomarer Modelle haben zahlreiche Forschungsteams diese Richtung kontinuierlich erforscht und eine Reihe von Fortschritten erzielt. Zum BeispielMit der vom Team der Universität Oxford entwickelten Methode des neuronalen Netzes nn-tm fcc lassen sich hochpräzise Potentialenergieflächenmodelle von Restfragmenten mit nahezu vollständiger quantenmechanischer Genauigkeit erstellen.Die mittleren quadratischen Fehler der Energie- und Atomkraftberechnungen liegen innerhalb von 1,0 kcal/mol bzw. 1,3 kcal/(mol·Å). Mit dieser Methode lassen sich Energie- und Atomkraftberechnungen für 15 repräsentative Proteine in nur 10 bis 100 Sekunden durchführen – tausendfach schneller als herkömmliche quantenmechanische Berechnungen.
Titel der Arbeit: Verbesserte Vorhersage von Proteinstrukturen mithilfe von Potenzialen aus dem Deep Learning
Link zum Artikel:https://www.nature.com/articles/s41586-019-1923-7
Ein weiteres deutsches Forschungsteam entwickelte den Quantenalgorithmus BF-DCQO, der eine nicht-variable iterative Strategie mit dem Ionenfallen-Quantencomputersystem IonQ kombiniert.Die Rechenzeit für ein 3D-Faltungsproblem mit 12 Aminosäuren konnte von 72 Stunden mit einem herkömmlichen GPU-Cluster auf etwa 4,3 Minuten reduziert werden.Die Geschwindigkeitssteigerung erreichte ebenfalls das Tausendfache.
Titel der Dissertation: Bias-Field-digitalisierter konterdiabatischer Quantenalgorithmus für binäre Optimierung höherer Ordnung
Link zum Artikel:https://www.nature.com/articles/s42005-025-02270-3
Insgesamt bietet die Kombination aus quantenmechanischen Methoden, Potenzialfunktionen des maschinellen Lernens und experimentellen Strukturdaten einen neuen technischen Ansatz zur Verfeinerung der Strukturen biologischer Makromoleküle und dürfte in Bereichen wie der Strukturmodellierung mit niedriger Auflösung, der Analyse von Ligandenbindungsmodi und der Erforschung funktioneller Zentren eine stabilere Rolle spielen.