Command Palette
Search for a command to run...
Die Grenzen Der Traditionellen Multimodalen Integration Werden Durchbrochen! Das MIT Präsentiert Das APOLLO-Framework, Das Eine Klare Trennung Von Zellübergreifenden Und Zellspezifischen Informationen ermöglicht.
In der Einzelzellbiologie erweitert die rasante Entwicklung von Messtechnologien stetig die Grenzen der wissenschaftlichen Forschung. Durchbrüche in Bereichen wie Multiplex-Bildgebung, Einzelzell-Transkriptomsequenzierung (scRNA-seq), Chromatin-Open-Sequencing (scATAC-seq) und Proteinmengenbestimmung ermöglichen es Forschern, einzelne Zellen umfassend und multidimensional zu beobachten, darunter Transkriptionsregulation, Chromatinstatus, Proteinexpression und morphologische Struktur. Diese multimodalen Daten interpretieren den Code des Lebens auf verschiedenen Ebenen, und ihre komplementäre Integration bietet eine beispiellose Möglichkeit, zelluläre Heterogenität aufzudecken und Krankheitsmechanismen zu erforschen.
Allerdings weisen die derzeitigen Analysemethoden bei der Verarbeitung solch hoher Datenmengen noch immer erhebliche Einschränkungen auf.Gängige Strategien beinhalten oft die separate Analyse jedes Modus und den anschließenden Vergleich, was nicht nur ineffizient ist, sondern es auch erschwert, die tiefen Korrelationen zwischen den Modi zu erfassen.Ein anderer Ansatz integriert multimodale Daten mittels Repräsentationslernen in denselben latenten Raum, verwechselt dabei aber oft gemeinsame Informationen mit modalitätsspezifischen Informationen und verschleiert so den einzigartigen Beitrag jeder Dimension zur Zellfunktion.
Dieses Problem tritt besonders deutlich bei der integrierten Analyse gepaarter scATAC-seq- und scRNA-seq-Daten auf.Herkömmliche Methoden differenzieren die Chromatinzugänglichkeit oft grob auf Genebene, um sie mit der Genexpression zu vergleichen. Dies vereinfacht zwar das Problem, kann aber feine Strukturinformationen auf Chromatinebene verwerfen und ist nur auf Datentypen mit relativ einheitlichen Eigenschaften anwendbar. Komplexere Integrationsmethoden wie lineare Modelle und generative adversarielle Netzwerke haben entweder Schwierigkeiten, sich an unstrukturierte Daten wie Bilddaten anzupassen, oder schneiden bei der Trennung von gemeinsamen und spezifischen Informationen schlecht ab und werden somit dem wachsenden Bedarf an multimodaler Datenanalyse in großen Biobanken nicht gerecht.
Angesichts der kontinuierlichen Weiterentwicklung der Einzelzelltechnologie und des rasanten Wachstums des Datenumfangs ist die effiziente und automatische Integration multimodaler Daten bei gleichzeitiger klarer Trennung gemeinsamer Informationen von modalitätsspezifischen Informationen zu einer zentralen Herausforderung der Einzelzellbiologie geworden.
Um dieser Herausforderung zu begegnen, hat ein gemeinsames Forschungsteam des MIT und der ETH Zürich ein allgemeines Deep-Learning-Computing-Framework namens APOLLO (Autoencoder with a Partially Overlapping Latent space learned through Latent Optimization) vorgeschlagen.Dieses Rahmenwerk bietet einen praktikablen technischen Weg für eine umfassendere und genauere Analyse von Zellzuständen und ihrer regulatorischen Logik, indem es gemeinsame Informationen und modalitätsspezifische Informationen explizit modelliert.
Die zugehörigen Forschungsergebnisse mit dem Titel „Partially shared multi-modal embedding learns holistic representation of cell state“ wurden in Nature Computational Science veröffentlicht.
Forschungshighlights:
* Diese Forschungsarbeit schlägt APOLLO vor, ein allgemeines Deep-Learning-Framework, das in multimodalen Daten automatisch und explizit "gemeinsame Informationen" von "modalitätsspezifischen Informationen" trennen kann.
* APOLLO lernt einen teilweise überlappenden latenten Raum, indem jede Modalität mit einem Autoencoder ausgestattet wird und eine zweistufige Trainingsstrategie angewendet wird. Dadurch werden biologische Signale, die üblicherweise über mehrere Modalitäten erfasst werden, effektiv identifiziert und unterschieden.
* APOLLO kann den Zusammenhang zwischen Unterschieden in der subzellulären Lokalisation von Proteinen und der Morphologie verschiedener Zellkompartimente aufzeigen und erweitert damit die Analyse von reinen Omics-Daten auf das Gebiet der räumlichen Morphologie.

Papieradresse:
https://www.nature.com/articles/s43588-025-00948-w
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „APOLLO“, um das vollständige PDF zu erhalten.
Datensatz: Umfassende Validierung einschließlich Sequenzierung und Bildgebung.
Um die Leistungsfähigkeit des APOLLO-Frameworks umfassend zu bewerten, nutzte die Studie mehrere öffentlich verfügbare multimodale Einzelzelldatensätze, die sowohl Sequenzierungs- als auch Bildgebungstechnologien abdecken.
Bezüglich der Sequenzierungsdaten,Die Forscher nutzten zunächst gepaarte Einzelzell-Transkriptomdaten (scRNA-seq) und Chromatinzugänglichkeitsdaten (scATAC-seq), die mit der SHARE-seq-Technologie gemessen wurden, um zu überprüfen, ob APOLLO automatisch Genaktivität identifizieren und unterscheiden kann, die sowohl durch Transkriptomdaten als auch durch Chromatinzugänglichkeitsdaten erfasst wird, sowie Genaktivität, die nur durch eine der beiden Modalitäten erfasst wird.
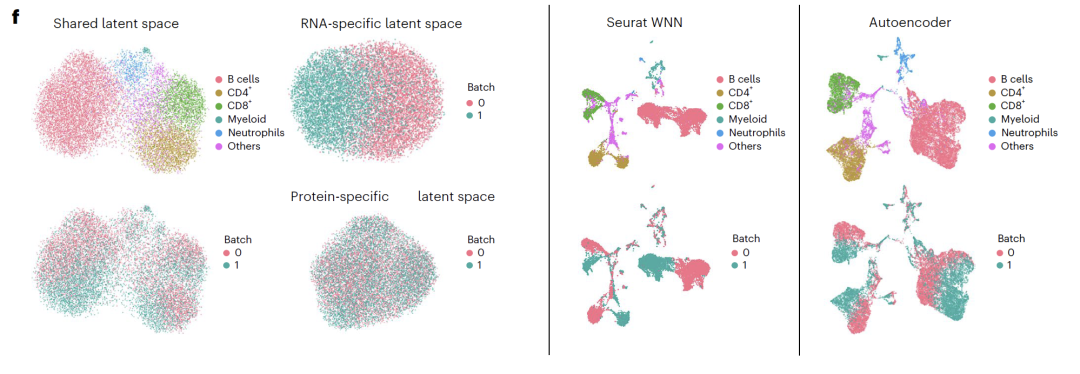
Zweitens verwendeten die Forscher gepaarte scRNA-seq- und Zelloberflächenprotein-Häufigkeitsdaten, die mittels CITE-seq gewonnen wurden, um die Anwendbarkeit des Modells auf Sequenzierungsdaten weiter zu testen.Der CITE-seq-Datensatz stammt aus Milz und Lymphknoten von Mäusen und umfasst zwei Gruppen von Wildtyp-Mausproben, die einer unabhängigen experimentellen Behandlung unterzogen wurden.Es kann nicht nur zur Beurteilung der Fähigkeit zur Zelltypunterscheidung verwendet werden, sondern deckt auch den experimentellen Batch-Effekt auf, der durch unterschiedliche Mausquellen verursacht wird.
Bezüglich der Bilddaten,Forscher präsentierten einen Multiplex-Bildgebungsdatensatz von humanen peripheren mononukleären Blutzellen (PBMCs). Dieser umfasst 32.345 Zellen von 40 Patienten, die in vier diagnostische Kategorien eingeteilt wurden: gesund, Meningeom, Gliom und Kopf-Hals-Tumoren. Von jedem Patienten wurden zwei Datensätze mit unterschiedlichen Antikörperkombinationen erhoben: Ein Datensatz verwendete DAPI zur Chromatinmarkierung in Kombination mit Antikörperfärbungen gegen CD4, CD8 und CD16; der andere Datensatz verwendete ebenfalls DAPI-Färbung, jedoch in Kombination mit Antikörperfärbungen gegen Lamin, CD3 und γH2AX.
Tests mit diesem Datensatz ergaben Folgendes:APOLLO kann Informationen über den Zellzustand identifizieren, die beiden Modalitäten in Bezug auf Chromatin-Struktur und Proteinlokalisierung gemeinsam sind, sowie morphologische Merkmale, die nur von einer einzigen Modalität erfasst werden.Darüber hinaus wurden durch die Kombination zusätzlicher Zellfärbemarker wie Mikrotubuli und endoplasmatisches Retikulum auch multiple Bildgebungsdaten aus dem Human Protein Atlas (HPA) verwendet, um zu zeigen, dass APOLLO dazu genutzt werden kann, den Zusammenhang zwischen Unterschieden in der subzellulären Lokalisation von Proteinen und der Morphologie verschiedener Zellkompartimente aufzudecken.
APOLLO-Modell: Ein Autoencoder, der eine latente Optimierungsstrategie verwendet
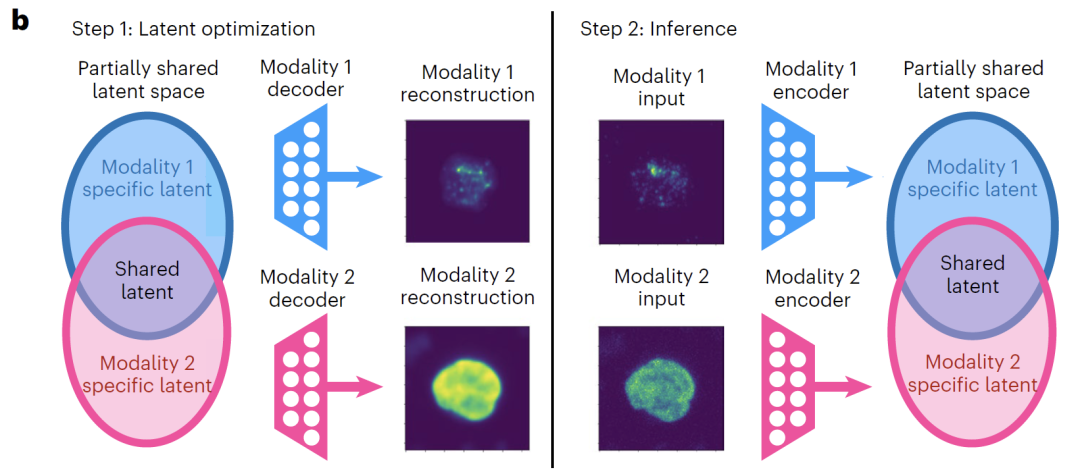
Um das häufige Problem bestehender multimodaler Integrationsmethoden zu lösen, die gemeinsame Informationen mit modalitätsspezifischen Informationen verwechseln, schlägt diese Studie das APOLLO-Framework vor. Dieses Framework nutzt latente Optimierung, um einen Autoencoder in einem teilweise überlappenden latenten Raum zu trainieren. Ziel ist es, gemeinsame und modalitätsspezifische Informationen über mehrere Modalitäten hinweg automatisch zu lernen und effektiv zu entkoppeln. Im Gegensatz zu herkömmlichen Autoencodern, die alle latenten Dimensionen einheitlich ausrichten,APOLLO führt die modalitätsübergreifende Ausrichtung nur auf einige der potenziellen Dimensionen durch und reserviert die übrigen Dimensionen für Informationen, die für jede Modalität spezifisch sind. Dadurch wird eine klare Trennung zwischen gemeinsamen und spezifischen Informationen im Modelldesign erreicht.
Im Hinblick auf die Modellarchitektur,APOLLO ist mit einem Autoencoder für jede Datenmodalität ausgestattet und kann je nach Bedarf zusätzliche Decoder einführen.Der Encoder und Decoder verwenden neuronale Netzwerkstrukturen, die an spezifische Modalitäten angepasst sind. So werden beispielsweise Faltungsnetzwerke für Bilddaten und vollständig verbundene Netzwerke für Genexpressionsdaten eingesetzt, um die Datencharakteristika jeder Modalität vollständig zu erfassen. Der latente Raum ist explizit in zwei Teile unterteilt: gemeinsame latente Merkmale und modalitätsspezifische latente Merkmale. Die Dimension des gemeinsamen latenten Raums ist üblicherweise deutlich größer als die des modalitätsspezifischen Raums, um eine ausreichende Repräsentation modalitätsübergreifender gemeinsamer Informationen zu gewährleisten.
Wie in der folgenden Abbildung dargestellt, besteht der Trainingsprozess von APOLLO aus zwei Schritten:Der erste Schritt konzentriert sich auf das Training der Decoder für jede Modalität, während gleichzeitig der latente Raum aktualisiert wird.Das Hauptziel besteht darin, dem Decoder die präzise Rekonstruktion der Eingangsdaten aus dem latenten Raum zu ermöglichen. Falls die Aufgabe eine stärkere Repräsentation gemeinsamer Informationen und eine modalitätsübergreifende Vorhersage erfordert, werden zwei zusätzliche Decoder eingeführt, die den gemeinsamen latenten Raum jeweils einer Modalität zuordnen. Das Training wird durch Minimierung des Rekonstruktionsverlusts abgeschlossen.
Der zweite Schritt besteht darin, den modalitätsspezifischen Encoder zu trainieren.Jeder Datenmodus wird seinem entsprechenden latenten Raum zugeordnet. Durch Minimierung des mittleren quadratischen Fehlers wird die Einbettung von Stichproben, die nicht am Training beteiligt waren, in den latenten Raum abgeleitet, wodurch sichergestellt wird, dass das Modell eine gute Generalisierungsfähigkeit aufweist.
Zur Validierung des Modells testete die Studie zunächst die Entkopplungsleistung von APOLLO an fünf simulierten Datensätzen mit bekannten realen zugrunde liegenden Strukturen.Die Ergebnisse zeigen, dass das Modell unabhängig von der Abhängigkeit zwischen gemeinsamen und spezifischen latenten Merkmalen eine stabile Leistungsfähigkeit aufrechterhalten kann.Weitere Validierungen anhand realer Daten zeigen, dass APOLLOs explizites Lernen der partiellen Informationsteilung nicht nur multimodale Informationen entkoppeln, sondern auch genaue crossmodale Vorhersagen ermöglichen kann, wie beispielsweise die Vorhersage unentdeckter Proteine aus der Chromatin-Bildgebung.
Insgesamt entkoppelt und interpretiert APOLLO effektiv gemeinsame und modalitätsspezifische Informationen in multimodalen Datensätzen, indem es teilweise gemeinsame latente Räume lernt und so einen allgemeinen Rahmen für die Aufdeckung biologischer Mechanismen bietet.
Über traditionelle multimodale Integrationsrahmen hinaus ist ein umfassenderes Verständnis zellulärer Zustände erforderlich.
Um die Universalität und die Kernvorteile des APOLLO-Modells umfassend zu bewerten, wurde eine Reihe von Experimenten in fünf Richtungen konzipiert: Integration von Paarsequenzierungsdaten, Integration von Chromatin- und Protein-Bildgebung, multimodale Vorhersage, Erkennung morphologischer Merkmale und Erforschung der subzellulären Lokalisierung von Proteinen.
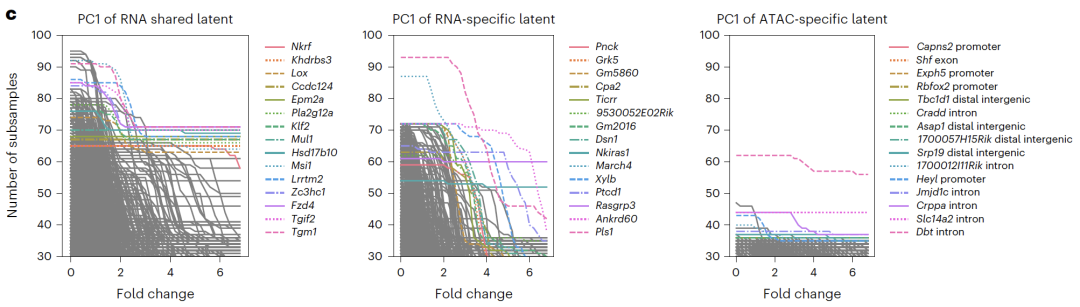
Bei der Integration von SequenzierungsdatenpaarenSHARE-seq-Experimente zeigten, dass die Hinzunahme eines modalitätsspezifischen Raums zum gemeinsamen Raum die Genauigkeit der Zelltypklassifizierung deutlich verbessern kann. Dies beweist, dass der spezifische Raum biologische Informationen erfassen kann, die im gemeinsamen Raum nicht enthalten sind.
Die potenzielle räumliche Interpretation ergab, dass RNA-spezifische Bereiche mit Zellzyklus-bezogenen Genen angereichert waren, ATAC-spezifische Bereiche mit offenen Chromatinregionen, die mit der Transkriptionsregulation in Zusammenhang stehen, und gemeinsame Bereiche mit bekannten Transkriptionsfaktoren und regulatorischen Signalwegen, was die biologische Bedeutung der Entkopplungsergebnisse bestätigte. In CITE-seq-ExperimentenAPOLLO trennte erfolgreich Zelltyp- und Batch-Effekte in einen gemeinsamen Raum und einen RNA-spezifischen Raum.Mit den bestehenden Integrationsmethoden lässt sich diese Art der Entkopplung nicht erreichen, was die einzigartigen Vorteile des Modells bei der Integration von Sequenzierungsdaten unterstreicht.
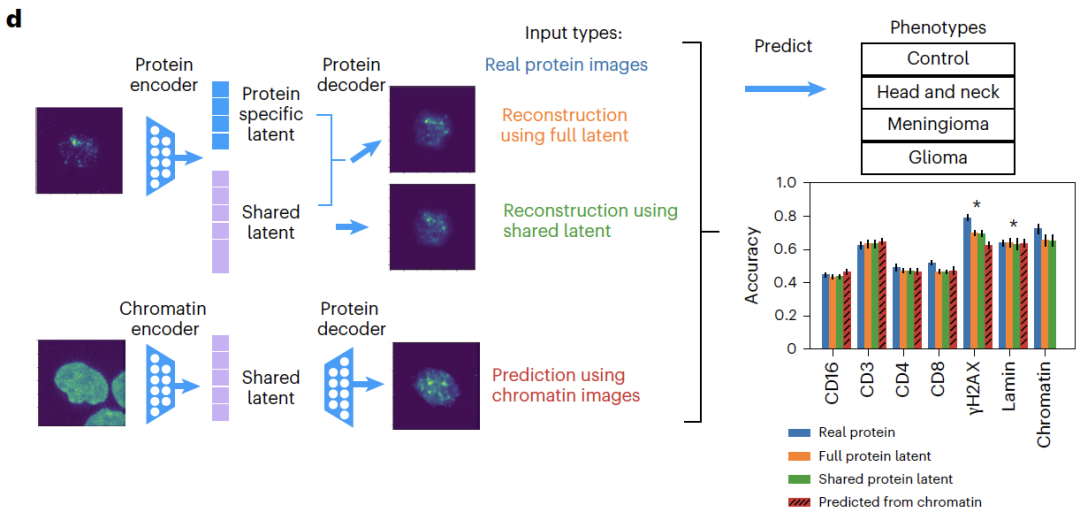
Bezüglich der Bilddaten,APOLLO kann Zellbilder von Patienten, die nicht an der Schulung teilgenommen haben, präzise rekonstruieren.Bei der modalitätsübergreifenden Aufgabe, nicht erkannte Proteine aus Chromatin vorherzusagen, ist APOLLO herkömmlichen Bildrekonstruktionsmethoden deutlich überlegen;Die nachfolgende phänotypische Klassifizierung zeigte, dass die auf der vorhergesagten Proteinbildgebung basierende Klassifizierungsgenauigkeit der Genauigkeit der tatsächlichen Bildgebung ähnelte, wobei das CD3-Protein die beste Vorhersageleistung aufwies. Dies bestätigt, dass die Vorhersageergebnisse effektiv für biologische Entdeckungen genutzt werden können.
Bei Aufgaben zur Erkennung morphologischer Merkmale,Der gemeinsame Bereich erfasst primär morphologische Merkmale des Chromatins (wie Kernfläche und Heterochromatinvolumen), während proteinspezifische Merkmale, wie die Anzahl fokaler γH2AX-Areale, nur in ihren jeweiligen spezifischen Bereichen existieren. Experimente zur Merkmalsablation zeigten, dass die Entfernung dieses Merkmals die Genauigkeit der phänotypischen Klassifizierung signifikant verringerte, was die Genauigkeit der Entkopplung weiter bestätigte.

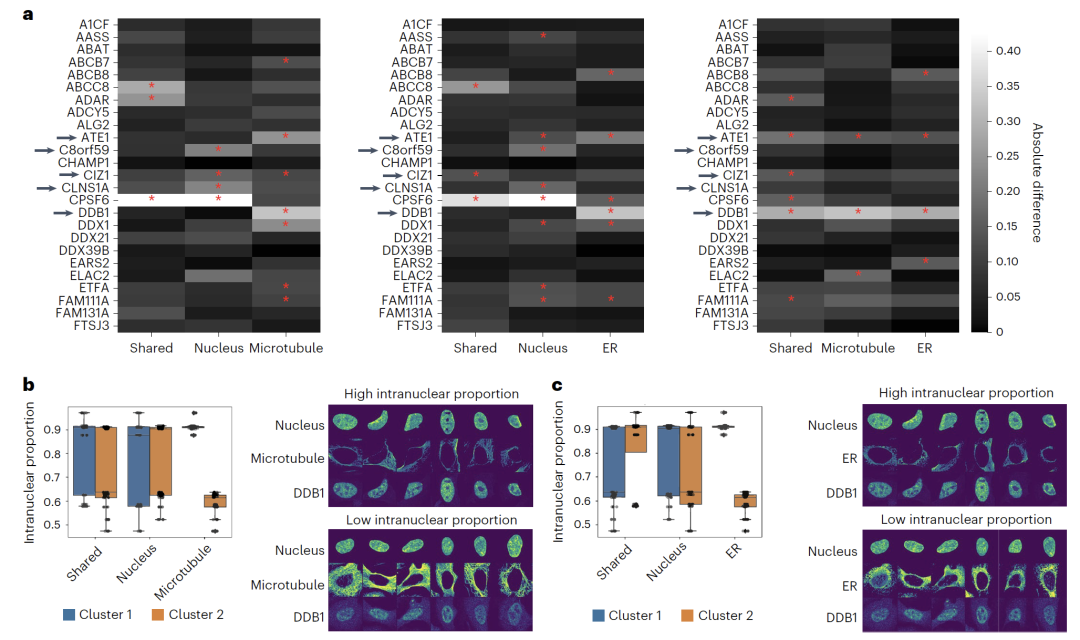
Bei der Untersuchung der subzellulären Lokalisierung von ProteinenDie Anwendung von APOLLO auf Bildgebungsdaten von U2OS-Zellen zeigte, dass Unterschiede in der Proteinlokalisierung innerhalb des Zellkerns durch Eigenschaften verschiedener Zellkompartimente erfasst werden können. Beispielsweise ist die nukleäre Lokalisierung von DDB1 mit dem endoplasmatischen Retikulum und der Mikrotubuli-Morphologie assoziiert, während CLNS1A ausschließlich mit der Kernmorphologie assoziiert ist. Dieses Ergebnis deutet darauf hin, dass…Das Modell kann auf verschiedene Bildgebungskombinationen erweitert werden und bietet so eine neue Perspektive zum Verständnis des Zusammenhangs zwischen Proteinlokalisierung und Zellmorphologie.
Implementierung der multimodalen Datenintegration einzelner Zellen
Die Integration multimodaler Einzelzelldaten entwickelt sich zu einer zentralen technologischen Richtung für die Analyse zellulärer Heterogenität, die Aufdeckung von Krankheitsmechanismen und die Förderung der Entwicklung der Präzisionsmedizin und hat in der globalen akademischen Gemeinschaft breite Aufmerksamkeit erregt.
Beispielsweise die scMTR-seq-Technologie, die vom Team um Peter Rugg-Gunn am Babraham Institute der Universität Cambridge entwickelt wurde...Zum ersten Mal wurde die gleichzeitige Erfassung von sechs Histonmodifikationen und des gesamten Transkriptoms auf Einzelzellebene erreicht.Sie überwanden einen technischen Engpass in der Epigenetikforschung, der ein Jahrzehnt lang bestanden hatte.
Titel des Papiers:
Kombinatorische Profilierung multipler Histonmodifikationen und des Transkriptoms in einzelnen Zellen mittels scMTR-seq
Link zum Artikel:
https://www.science.org/doi/10.1126/sciadv.adu3308
Das vom Forschungsteam der Stanford University vorgeschlagene CellFuse-Framework konstruiert einen gemeinsamen Einbettungsraum auf Basis von überwachtem kontrastivem Lernen und ist speziell für multimodale Integrationsszenarien mit begrenzter Merkmalsüberlappung konzipiert.Es ermöglicht eine präzise Zelltypvorhersage und eine nahtlose Integration über verschiedene Modalitäten und Versuchsbedingungen hinweg.Tests an mehreren Datensätzen, darunter gesunde PBMCs, Knochenmark, CAR-T-Therapie bei Lymphomen und Tumorgewebe, zeigen, dass das Framework bestehende Methoden sowohl hinsichtlich der Integrationsqualität als auch der operativen Effizienz übertrifft.
Titel des Papiers:
CellFuse ermöglicht die multimodale Integration von Einzelzell- und räumlichen Proteomikdaten.
Link zum Artikel:
Führende globale Biotechnologie- und Gesundheitsunternehmen beschleunigen den Einsatz von Technologien zur multimodalen Datenintegration einzelner Zellen. Ihr Fokus liegt dabei auf zentralen Anwendungsbereichen wie der klinischen Translation, der Arzneimittelentwicklung und der Präzisionsmedizin, um Spitzenforschung in die Praxis umzusetzen. Das deutsche Unternehmen BioNTech nutzt diese Technologie für die Tumorimmuntherapie und die Entwicklung personalisierter Impfstoffe. Durch die Integration von Einzelzell-RNA-Sequenzierung, Proteinexpressionsprofilierung und räumlichen Transkriptomikdaten analysiert BioNTech präzise die zelluläre Heterogenität im Tumormikromilieu, identifiziert wichtige Immunzellsubtypen und zugehörige Biomarker und liefert so wichtige Daten für die Entwicklung und Optimierung personalisierter Tumorimpfstoffe. Dadurch werden die Zielgenauigkeit und Wirksamkeit der Impfstoffe signifikant verbessert.
Es ist absehbar, dass die Entschlüsselung des Lebens auf Einzelzellebene, angetrieben durch kontinuierliche Durchbrüche in der multimodalen Integrationstechnologie, schließlich von der Vision zur Realität werden und der Präzisionsmedizin der Zukunft neuen Schwung verleihen wird.