Command Palette
Search for a command to run...
Einem Französischen Team Gelang Es, 2,39 Millionen Antiphagenproteine Vorherzusagen Und Mithilfe Eines Deep-Learning-Modells Die Antivirale Immunität Von Bakterien abzubilden.

In der mikroskopischen Welt ist das „Wettrüsten“ zwischen Bakterien und Bakteriophagen nie erloschen. Bakteriophagen sind Bakterien in der Regel um etwa das Zehnfache überlegen und nutzen Bakterien als Wirte für ihre eigene Vermehrung. Gleichzeitig haben Bakterien im Laufe der Evolution hochdiverse antivirale Abwehrsysteme entwickelt. Aktuell sind über 250 Anti-Phagen-Systeme experimentell validiert, darunter verschiedene Mechanismen wie Restriktions-Modifikationssysteme und CRISPR-Cas-Systeme, und ständig werden neue Systeme entdeckt. Dieses Phänomen deutet darauf hin, dass die Komplexität und Vielfalt bakterieller Abwehrsysteme unser derzeitiges Verständnis bei Weitem übersteigt.Aufgrund der Beschränkungen traditioneller experimenteller Methoden und computergestützter Verfahren bleibt eine große Anzahl potenzieller Anti-Phagen-Mechanismen im bakteriellen Genom verborgen und wurde noch nicht systematisch erforscht.
Bisherige Forschungsergebnisse haben bestimmte Gemeinsamkeiten bekannter Antiphagensysteme auf Ebene der Proteinsequenz und der Genomorganisation festgestellt, wie beispielsweise das wiederkehrende Auftreten charakteristischer Domänen und deren gehäufte Verteilung in „Verteidigungsinseln“ oder Präphagenregionen. Diese Muster legen Folgendes nahe:Wenn diese gemeinsamen Muster identifiziert und genutzt werden können, ist es möglicherweise möglich, unbekannte Antiphagensysteme systematisch auf der Ebene des gesamten Genoms aufzudecken.
Aufbauend auf diesem Ansatz entwickelten und optimierten Forscher des Pasteur-Instituts in Frankreich drei komplementäre Deep-Learning-Modelle zur großflächigen Vorhersage von Phagenresistenz. Das ALBERT_DF-Modell stützt sich ausschließlich auf den lokalen genomischen Kontext für seine Schlussfolgerungen; ESM_DF verwendet ein Protein-Sprachmodell zur Analyse von Aminosäuresequenzen; und GeneCLR_DF integriert Sequenzinformationen mit dem genomischen Kontext. In einem einheitlichen Benchmark-TestGeneCLR_DF erzielte die besten Ergebnisse mit einer Präzision von 991 TP3T und einem Recall von 921 TP3T.
Aufbauend auf diesem hochpräzisen Modell führte die Studie zudem genomweite Vorhersagen von Antiphagensystemen durch. Die Ergebnisse zeigten, dass in über 32.000 Bakteriengenomen etwa 1,51 TP3T-Gene in einem typischen Bakteriengenom an der antiviralen Abwehr beteiligt sind; noch wichtiger ist, dass über 851 TP3T-Gene, die eine vorhergesagte, abwehrrelevante Proteinfamilie repräsentieren, noch nie zuvor mit Immunfunktionen in Verbindung gebracht wurden.Das Modell sagte ungefähr 2,39 Millionen Antiphagenproteine voraus, von denen eine große Anzahl zu Einzelgen-Abwehrsystemen gehört, und definierte ungefähr 23.000 Operonfamilien auf der Grundlage von Gen-Kookkurrenzbeziehungen.Die überwiegende Mehrheit dieser Bakterien stand bisher in keinem Zusammenhang mit der antiviralen Abwehr. Zusammengenommen zeichnen diese Ergebnisse ein systematisches Bild der bakteriellen antiviralen Immunität und offenbaren deren Ausmaß und Vielfalt, die den bisherigen Kenntnisstand weit übertrifft.
Die zugehörigen Forschungsergebnisse mit dem Titel „Protein- und genomische Sprachmodelle enthüllen die unerforschte Vielfalt der bakteriellen Immunität“ wurden in Science veröffentlicht.
Forschungshighlights:
* Insgesamt wurden 2,39 Millionen Antiphagenproteine vorhergesagt, von denen 85% noch nie zuvor mit einer Immunfunktion in Verbindung gebracht worden war;
* In einem typischen Bakteriengenom sind etwa 1,51 TP3T-Gene spezifisch für die antivirale Abwehr verantwortlich.
Es wurden etwa 23.000 Manipulator-Unterfamilien vorhergesagt, von denen die überwiegende Mehrheit zum ersten Mal entdeckt wurde;
* Eine große Anzahl vorhergesagter Abwehrproteine existiert in Form von Einzelgensystemen, was die herkömmliche Ansicht in Frage stellt, dass Abwehrfunktionen üblicherweise durch das Zusammenwirken mehrerer Gene erreicht werden.

Papieradresse:
https://www.science.org/doi/10.1126/science.adv8275
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „GeneCLR“, um das vollständige PDF zu erhalten.
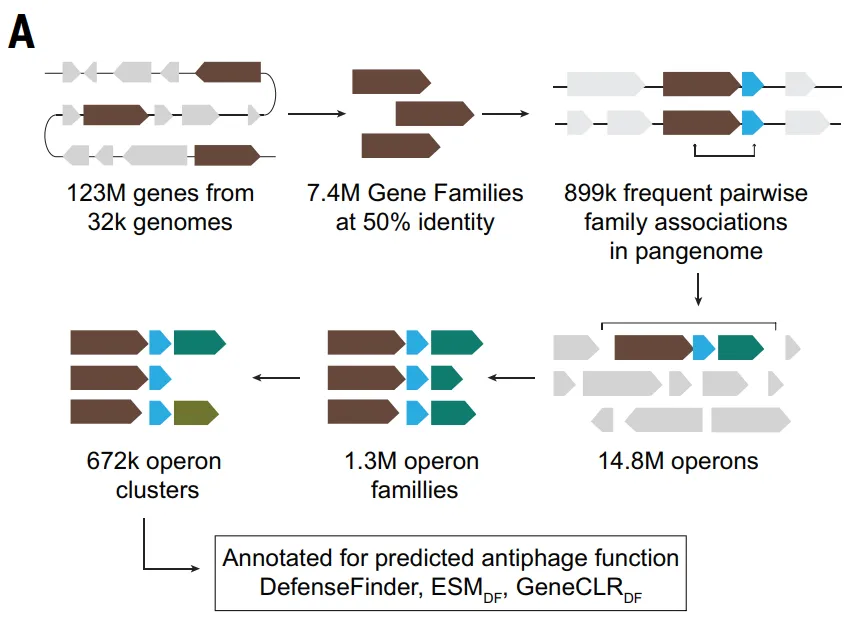
Datensatz: Basierend auf 123 Millionen Proteinen und 32.000 Genomen
In dieser Studie wurden zunächst die Tools DefenseFinder und PadLoc verwendet.Um bekannte Antiphagensysteme quantitativ zu charakterisieren, wurde ein systematischer Scan von 32.798 vollständigen Bakteriengenomen in der RefSeq-Datenbank durchgeführt.Von den ungefähr 123 Millionen Proteinen identifizierte DefenseFinder v1.3 521.360, was 0,41 TP3T entspricht und zu den Komponenten des Antiphagensystems gehört, während PadLoc 805.357 identifizierte, was 0,651 TP3T entspricht.
Es ist bemerkenswert, dass viele Abwehrsysteme ursprünglich durch genomische Assoziationen mit bekannten Systemen entdeckt wurden. Diese Assoziationen lassen sich auf Ebene der Proteinfamilien mithilfe eines „Abwehr-Scores“ quantifizieren, der die Häufigkeit misst, mit der eine bestimmte Proteinfamilie im Genom gemeinsam mit bekannten Abwehrproteinen vorkommt.
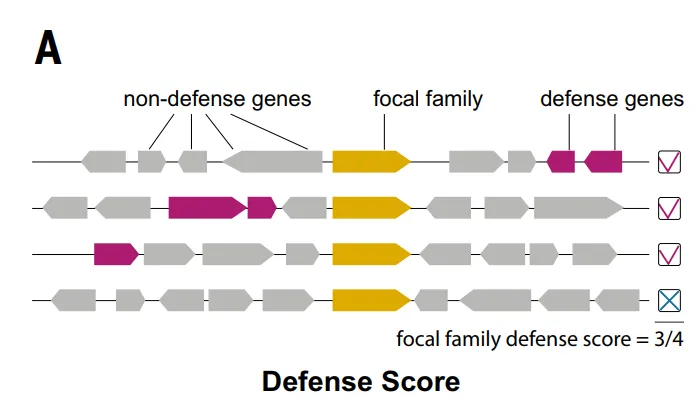
Basierend auf der Verteidigungswertungsmethode, wie in der folgenden Abbildung dargestellt.Die Forscher identifizierten insgesamt 37.959 Proteinfamilien (4,61 % von TP3T) als Kandidaten für Antiphagenfamilien.Anschließend wurden 7.799 Familien, wie zum Beispiel Integrasen, die mit zentralen biologischen Funktionen oder mobilen genetischen Elementen in Verbindung standen, ausgeschlossen, sodass schließlich 30.160 ausgewählte Kandidatenfamilien übrig blieben (was 3,71 TP3T entspricht).
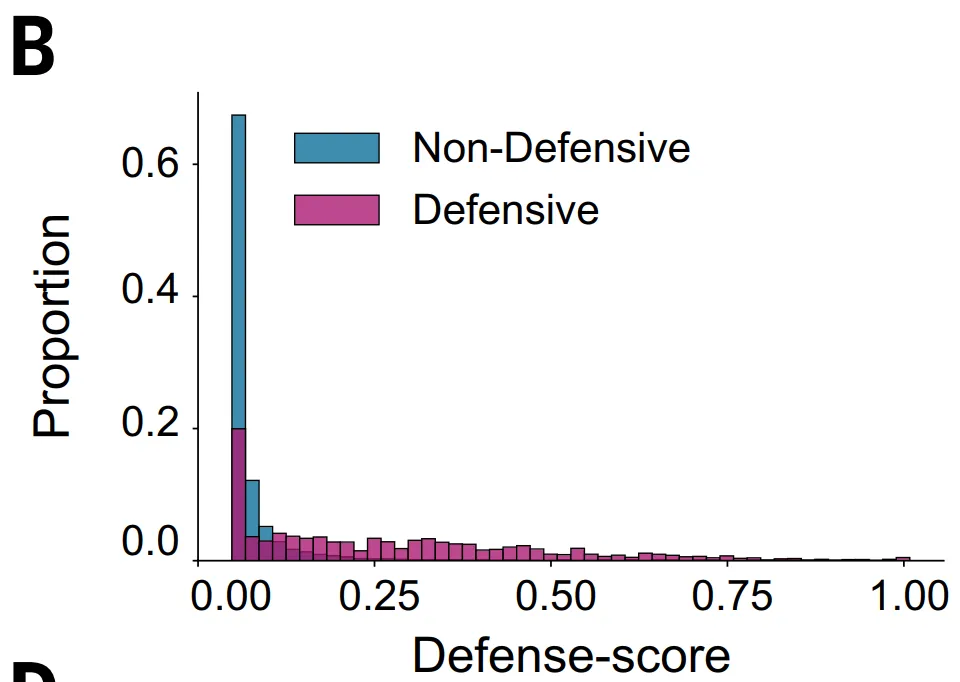
Diese Methode hat jedoch offensichtliche Einschränkungen:Erstens,Dies gilt nur für Proteinfamilien mit mehr als fünf homologen Sequenzen, wodurch Proteine mit etwa 23% ausgeschlossen werden.Zweitens,Manche Antiphagensysteme befinden sich nicht in typischen Abwehrinseln, und selbst wenn sie Abwehrfunktionen haben, können ihre Abwehrwerte niedrig sein, sodass sie übersehen werden.
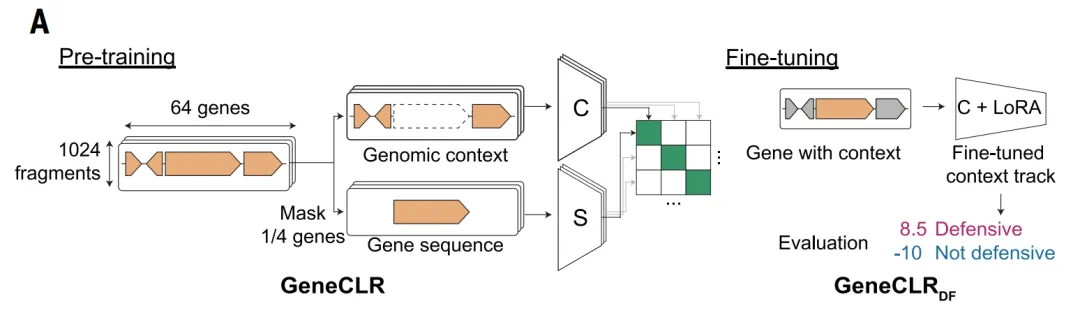
Um die oben genannten Einschränkungen zu überwinden und verteidigungsrelevante genomische Signale umfassender zu erfassen,Die Studie erstellte außerdem einen für Deep Learning geeigneten Datensatz.Im Rahmen des ALBERT_DF-Modells wurde das bakterielle Genom auf eine „linguistische“ Weise modelliert: Jede Proteinfamilie wurde als „Wort“ und benachbarte Gensegmente als „Satz“ behandelt.
Da der vollständige Datensatz über 8 Millionen verschiedene Proteinfamilien umfasst und damit den Wortschatz traditioneller Sprachmodelle bei Weitem übersteigt,Die Studie beschränkte den Trainingsumfang auf den Stamm der Actinobacteria und erstellte einen Datensatz mit 10.796 Genomen.Die Gene wurden in 4,2 Millionen Proteinfamilien gruppiert, wobei das Vokabular auf die 524.288 häufigsten Familien beschränkt war, wodurch ungefähr 891 TP3T-Proteine abgedeckt wurden.
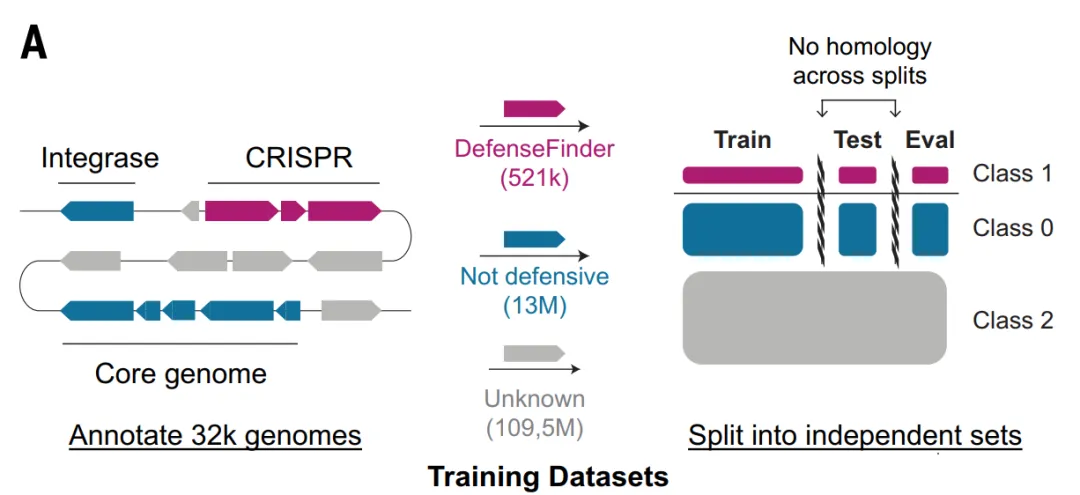
Für die Modelle ESM_DF und GeneCLR_DF wurde der Datensatz Gembase_DF erstellt: Wie in der folgenden Abbildung dargestellt, wurden 521.360 mit DefenseFinder markierte Antiphagenproteine als positive Proben verwendet, 116 Millionen hochkonservierte Kerngene, die in mehr als 99% vorkommen, und 14 Millionen nicht-defensive mobile genetische Elemente wurden als negative Proben verwendet, und die restlichen Proteine wurden als unmarkierte Kandidaten beibehalten.
Um Informationslecks zwischen Training, Validierung und Test zu vermeiden, wurden in der Studie alle Proteine desselben Abwehrsystems in denselben Datenfold gruppiert und MMseqs2 verwendet, um verbleibende Homologien über die Datenfolds hinweg zu entfernen und so die Strenge der Modellevaluierung zu gewährleisten.
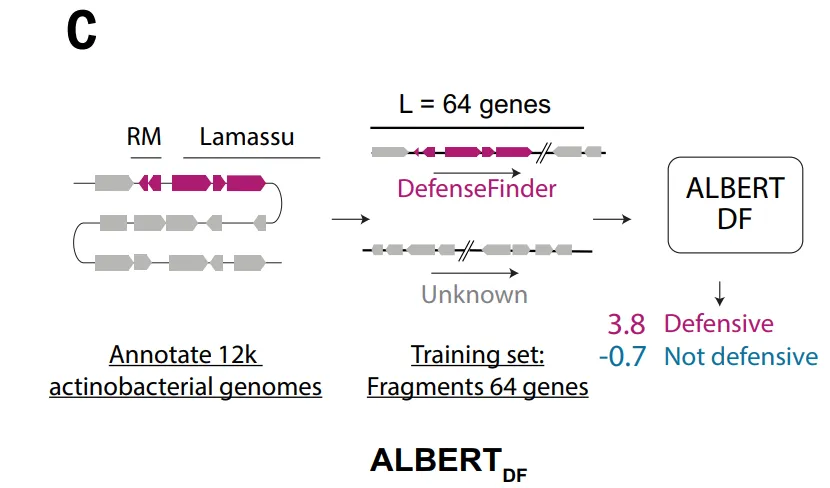
Modellarchitektur: Ein dreischichtiges Deep-Learning-Modell, das schrittweise vorgeht.
Um die Einschränkungen der traditionellen „Verteidigungsscore“-Methoden zu überwinden, entwickelte das Forschungsteam ein komplementäres und progressives Deep-Learning-Framework mit dem Ziel, drei Ziele zu erreichen: die Entdeckung unbekannter Systeme, die Analyse des gesamten Genoms und die hochpräzise integrierte Vorhersage.Im Einzelnen umfasst dies ALBERT_DF basierend auf dem genomischen Kontext, ESM_DF basierend auf der Proteinsequenz und GeneCLR_DF, das Sequenz- und Kontextinformationen integriert.
ALBERT_DF konzentriert sich dabei auf das Lernen funktioneller Signale aus Gen-"Nachbarschaftsbeziehungen" und ist in der Lage, neuartige Abwehrsysteme zu entdecken; ESM_DF verwendet direkt die Aminosäuresequenzmodellierung und verfügt über eine gute sequenzübergreifende Generalisierungsfähigkeit; GeneCLR_DF integriert die beiden Informationstypen in einem einheitlichen Rahmen und erzielt ein besseres Gleichgewicht zwischen Erkennungsgenauigkeit und Vorhersageabdeckung.
Das ALBERT_DF-Modell basiert auf einer zentralen Beobachtung: Antiphagensysteme neigen dazu, im Genom geclustert vorzukommen, wobei stabile Organisationsmuster innerhalb und zwischen benachbarten Genen existieren. Ausgehend von dieser Eigenschaft,Diese Studie führt die ALBERT-Architektur aus der natürlichen Sprachverarbeitung in die Genommodellierung ein.Indem wir Proteinfamilien als „Wörter“ und Gensequenzen als „syntaktische Strukturen“ betrachten, lernen wir den lokalen Kontext durch die Vorhersage maskierter Gene kennen.
Im Gegensatz zu herkömmlichen, auf Sequenzähnlichkeit basierenden Methoden nutzt dieser Modellierungsansatz direkt Informationen zur genomischen Organisation und bietet daher ein größeres Potenzial zur Identifizierung neuartiger Abwehrmechanismen, die keine Homologie zu bekannten Systemen aufweisen. Aufgrund seiner Abhängigkeit von diskretisierten „lexikalischen“ Repräsentationen stößt diese Methode jedoch bei der Anwendung auf andere Spezies an ihre Grenzen.
Das ESM_DF-Modell hingegen verfolgt einen anderen Ansatz und wirkt direkt auf die Aminosäuresequenz des Proteins ein.Dieses Modell lernt die Kovariationen zwischen Resten und die Beziehungen zwischen Sequenzen über größere Distanzen durch umfangreiches Vortraining.Dies ermöglicht die Extraktion funktioneller Signale ohne künstliche Merkmale. Nach der Feinabstimmung kann ESM_DF jedes Protein bewerten, um festzustellen, ob es an der Phagenabwehr beteiligt ist. Dieser Ansatz verbessert die Anwendbarkeit der Methode deutlich und ermöglicht ihre Anwendung im Pan-Genom-Maßstab. Gleichzeitig hängt die Unterscheidungsfähigkeit von ESM_DF jedoch weiterhin teilweise von der Sequenzähnlichkeit ab. Daher eignet es sich besser zur Identifizierung entfernter Varianten bekannter Abwehrsysteme, während die Fähigkeit zur Identifizierung neuer Domänen ohne Homologie relativ begrenzt ist.
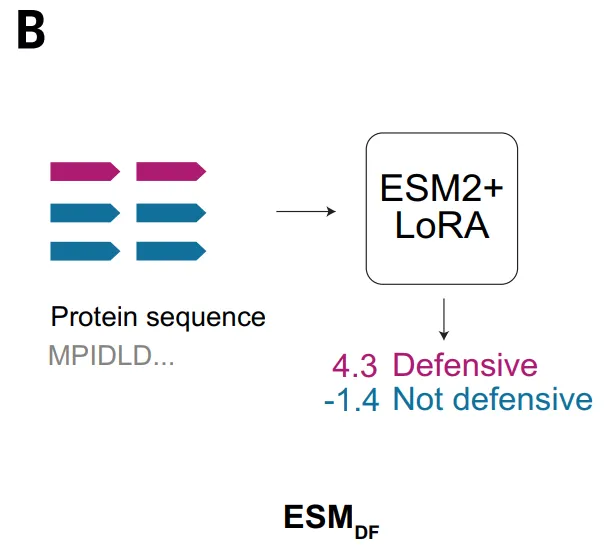
Auf dieser Grundlage wurde das GeneCLR_DF-Modell vorgeschlagen, um Sequenz- und genomische Kontextinformationen zu integrieren.Dieses Modell verwendet ein kontrastives Lernverfahren, bei dem gleichzeitig zwei Repräsentationen für jedes Gen gelernt werden:Eine Repräsentationsart basiert auf der Proteinsequenz, die andere auf ihrer genomischen Umgebung. Durch das Training des Modells wird ermittelt, ob diese beiden Repräsentationen demselben Gen entsprechen, wodurch die beiden Informationstypen im Repräsentationsraum aufeinander abgestimmt werden.
Dieses Design bietet einen entscheidenden Vorteil: Wenn bestimmte Gene auf Sequenzebene keine Homologie aufweisen, kann ihr typischer genomischer Kontext dennoch Hinweise zur Identifizierung liefern; umgekehrt können Sequenzmerkmale auch dann die Unterscheidung unterstützen, wenn die Kontextinformationen atypisch sind. Durch diesen komplementären MechanismusGeneCLR vereint die Fähigkeit, neuartige Systeme zu entdecken, mit der Skalierbarkeit für groß angelegte Anwendungen bei nachfolgenden Vorhersagen.
Insgesamt bilden diese drei Modelltypen einen klaren technischen Pfad: vom kontextbasierten lokalen Musterlernen über die sequenzbasierte globale Generalisierung bis hin zur einheitlichen Modellierung von Informationen aus verschiedenen Quellen. Dieser hierarchische Aufbau vermeidet nicht nur die Einschränkungen einer einzelnen Methode, sondern bietet auch einen universelleren technischen Rahmen für die systematische Erforschung unbekannter Antiphagenmechanismen.
Erreichen Sie eine Präzision von 991 TP3T und eine Trefferquote von 921 TP3T.
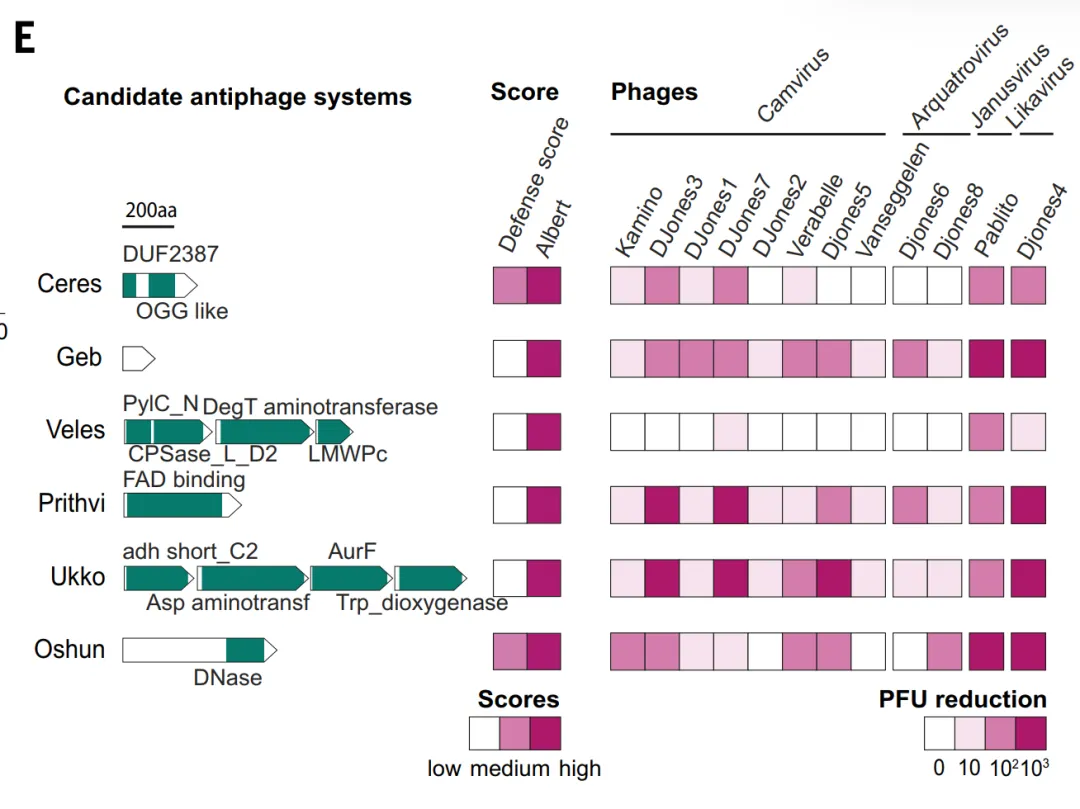
In der experimentellen Validierung wurde zunächst die Vorhersagekraft von ALBERT_DF evaluiert.Das Modell sagte insgesamt 1.930 Kandidaten für antiphage Proteinfamilien voraus, von denen etwa 331 TP3T mit den Ergebnissen der Verteidigungsscore-Methode übereinstimmten.Forscher wählten zehn Kandidatensysteme aus, denen sowohl Abwehrscores als auch bekannte Homologien fehlten, exprimierten diese in *Streptomyces whiteus* und testeten sie mit zwölf Phagen. Sechs dieser Systeme zeigten einen starken Schutz und reduzierten die Plaque-bildenden Einheiten um mehr als das Hundertfache. Diese Systeme (wie Ceres und Geb) enthalten Stoffwechselenzyme und kleine Proteine mit unbekannten Funktionen, die über den Rahmen klassischer Abwehrdomänen hinausgehen. Dies beweist, dass genomische Kontext-basierte Methoden neuartige Abwehrmechanismen aufdecken können, die mit traditionellen Methoden schwer zu identifizieren sind.
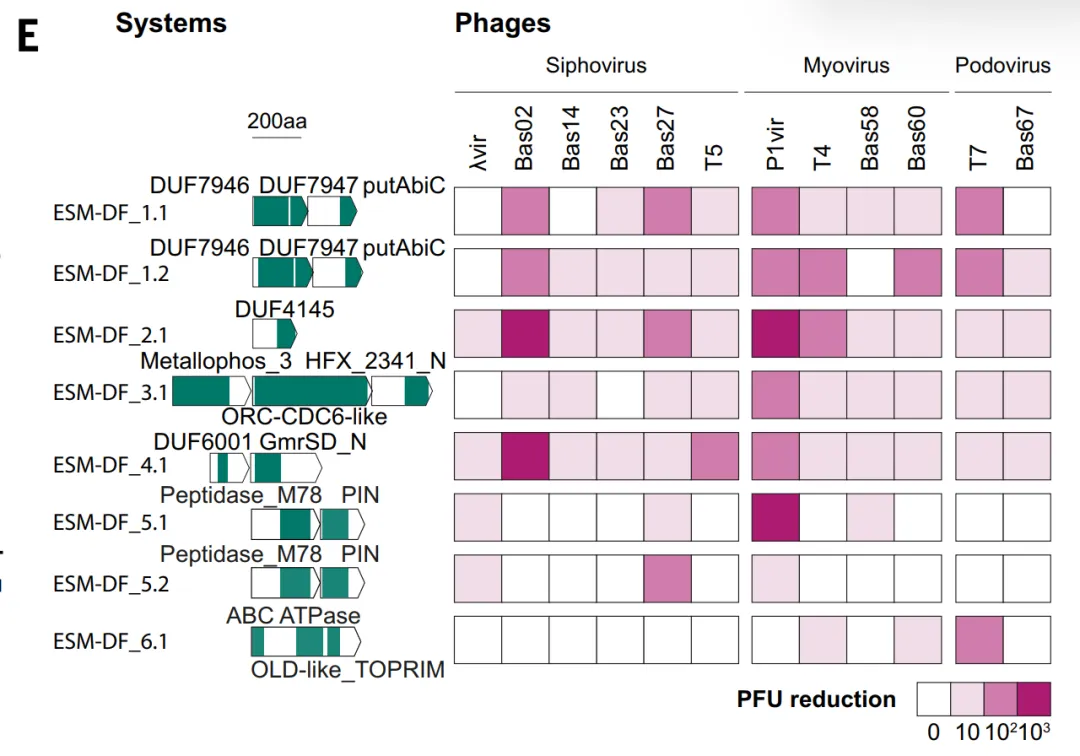
Im Rahmen der Validierung von ESM_DF wurde eine Gruppe vielversprechender Kandidaten in E. coli getestet. Sechs dieser Systeme, darunter ESM_DF, das gegen verschiedene Bakteriophagentypen resistent ist, zeigten antiphagische Eigenschaften. Zu diesen Systemen gehörten Varianten bekannter Abwehrdomänen sowie Domänen, die bisher nicht mit antiphagischer Funktion in Verbindung gebracht wurden, wie beispielsweise DUF7946.Dies deutet darauf hin, dass ESM nicht nur auf Sequenzhomologie beruht, sondern auch ein breiteres Spektrum funktioneller Merkmale identifizieren kann, aber insgesamt tendiert es immer noch dazu, eine Erweiterung bekannter Systeme zu sein.
GeneCLR_DF schnitt bei der Systemevaluierung am besten ab. Auf dem TestdatensatzDie Vorhersagewerte ermöglichen eine klare Unterscheidung zwischen defensiven und nicht-defensiven Proteinen.In der evolutionären Analyse wurden wichtigen Verteidigungszweigen wie Retrotranskriptoren, CBASS und Thoeris durchweg hohe Punktzahlen zugewiesen, während ESM-650M_DF diese nur teilweise identifizieren konnte.
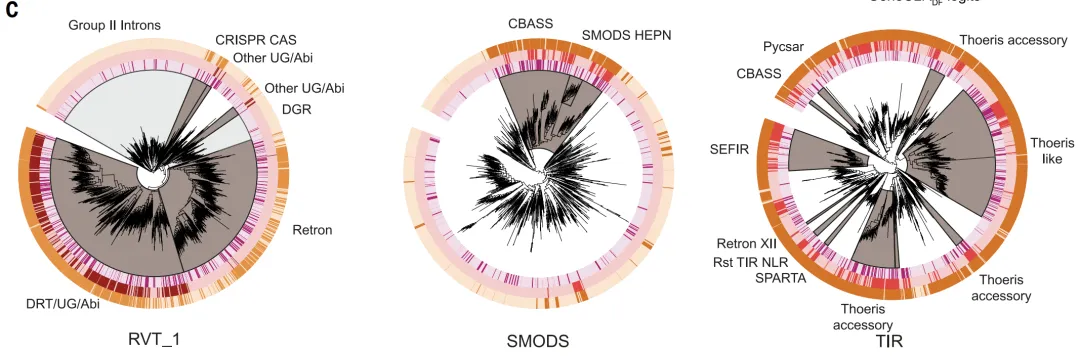
In unterschiedlichen genomischen Kontexten (Verteidigungsinseln, Integrone, Präphagenregionen),GeneCLR_DF kann das Verteidigungsmodul genau lokalisieren.Die quantitativen Ergebnisse zeigten, dass GeneCLR_DF bei einem Schwellenwert von −0,74 eine Präzision von 991 TP3T und eine Sensitivität von 92,41 TP3T erreichte; ESM_DF erkannte bei gleicher Präzision nur 581 TP3T. Mit einer Falsch-Positiv-Rate von 11 TP3T identifizierte GeneCLR_DF 941 TP3T in bekannten Abwehrproteinfamilien, signifikant mehr als ESM-650MDF (351 TP3T) und die Methode der Abwehrfraktion (51 TP3T). GeneCLR_DF identifizierte lediglich 561 TP3T-Familien und erkannte zudem 751 TP3T aus den 110 neu hinzugefügten Systemen. Von den 615.672 Kandidatenproteinfamilien wurden 931 TP3T ausschließlich von GeneCLR_DF detektiert.
Auf Operon-Ebene ergab eine weiterführende Analyse mittels kollinearer Clusterbildung, dass zahlreiche Abwehrstrukturen weiterhin unbekannt sind: Die vorhergesagte Proteinfamilie von 85% wurde lediglich durch ESM_DF und GeneCLR_DF identifiziert, während der Operon-Familie von 45% und dem Operon-Cluster von 52.7% bisher funktionelle Annotationen fehlten. Evolutionsanalysen zeigten zudem, dass…Der Mediananteil der Abwehrgene im bakteriellen Genom stieg von 0,46% auf 1,53%.Darüber hinaus sind zahlreiche Systeme mit mobilen genetischen Elementen angereichert, wobei sich 23.5% innerhalb der MGE-Grenze befindet und Satellitenelemente von 47.1% vermutlich Verteidigungsfähigkeiten kodieren.
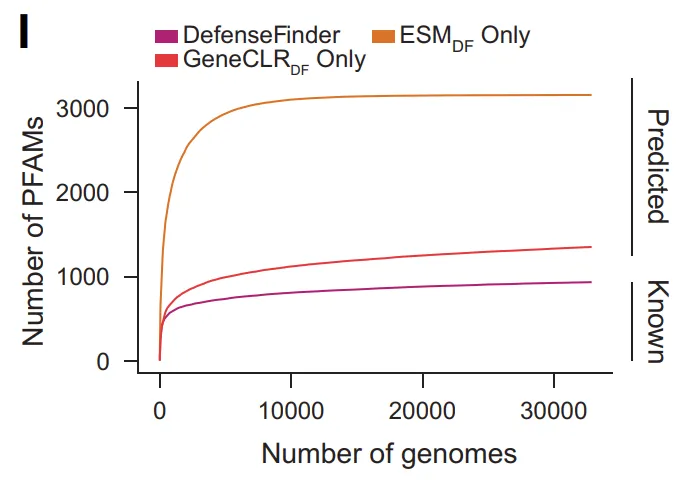
Auf der Ebene der molekularen Diversität erweiterte GeneCLR_DF die Anzahl der abwehrrelevanten Pfam-Familien von 934 auf 3.154 (etwa 15% aller Pfams). Gleichzeitig fehlten über 400.000 vorhergesagten Proteinfamilien jegliche Pfam-Annotationen, wobei weniger als 5% in DefenseFinder aufgeführt waren; über 3.500 Operon-Familien bestanden ausschließlich aus Proteinen ohne bekannte Domänen. Diese Ergebnisse deuten darauf hin, dass…Ein Großteil des molekularen Spektrums der Antiphagenabwehr ist noch nicht systematisch charakterisiert worden.
Deep Learning ermöglicht einen Quantensprung in der Effizienz der Entdeckung von Abwehrmechanismen gegen Phagen.
Vorhersagemodelle für antiphagische Systeme auf Basis von Deep Learning und die darauf aufbauenden antiviralen Immunatlanten bakterieller Organismen eröffnen einen skalierbareren Forschungsweg in diesem Bereich: weg von punktuellen Durchbrüchen, die auf Einzelfallstudien beruhen, hin zu systematischer Analyse mittels Mustererkennung. Dieser Wandel verbessert nicht nur die Effizienz der Entdeckung neuer Abwehrmechanismen, sondern führt auch zu einer engeren Verzahnung von akademischer Forschung und industriellen Anwendungen.
In der akademischen Forschung hat sich dieser Ansatz rasant verbreitet. Zahlreiche Forschungseinrichtungen kombinieren maschinelles Lernen mit Genomanalysen, um phagenresistente Systeme in größerem Umfang zu identifizieren. Zum BeispielDas DefensePredictor-Modell, entwickelt von einem Team am MIT,Durch die Anwendung der Modellierungslogik von Protein-Sprachmodellen und die Integration von Gensequenz- und genomischen Kontextinformationen konnte eine hochsensitive Identifizierung von Antiphagenproteinen erreicht werden. Das Modell wurde anhand von ca. 17.000 prokaryotischen Referenzgenomen trainiert und identifizierte in unabhängigen Tests ca. 821 neue TP3T-Abwehrsysteme, wodurch die Machbarkeit der „musterbasierten Entdeckung unbekannter Funktionen“ weiter bestätigt wurde.
Titel der Veröffentlichung: DefensePredictor: Ein maschinelles Lernmodell zur Entdeckung prokaryotischer Immunsysteme
Link zum Artikel:
https://www.science.org/doi/10.1126/science.adv7924
In der Industrie werden verwandte Technologien ebenfalls rasch implementiert. Angesichts der zunehmenden Antibiotikaresistenz gewinnen Bakteriophagen und ihre Folgetechnologien wieder an Bedeutung und werden zu einem entscheidenden Ansatzpunkt für den Ersatz oder die Ergänzung traditioneller Antibiotika. Locus Biosciences, ein Unternehmen in der klinischen Entwicklungsphase, hat eine Plattform auf Basis gentechnisch veränderter Bakteriophagen entwickelt. Durch die Kombination von maschinellem Lernen und synthetischer Biologie wurde LBP-EC01, ein Therapieansatz gegen multiresistente E. coli, entwickelt und damit die Präzision und Kontrollierbarkeit der Phagentherapie verbessert.
Micreos verfolgt hingegen einen anwendungsorientierten Ansatz und konzentriert sich auf die Industrialisierung von Bakteriophagen und Endosomalinen. Das Produkt Listex wird in der Lebensmittelverarbeitung zur Hemmung von Listerien-Kontaminationen eingesetzt und hat in mehreren Ländern Zulassungen erhalten; Staph Efekt nutzt die spezifischen bakteriziden Eigenschaften von Endosomalinen in der Hautpflege. Dieser Ansatz betont die „funktionale Umsetzung“ – die Überführung von Antiphagen-Mechanismen in konkrete, anwendbare Produkte, anstatt auf Laborebene zu verharren.
Insgesamt entwickelt sich die Antiphagenforschung von algorithmischen Modellen über experimentelle Überprüfung bis hin zu industriellen Anwendungen immer weiter zu einer geschlosseneren Kette. Es ist absehbar, dass dieser Weg – beginnend mit Berechnungen, verifiziert durch Experimente und geleitet von Anwendungen – mit zunehmender Datenmenge und der Iteration von Modellen weiterhin zu einem tieferen Verständnis bakterieller Immunsysteme beitragen und diese Erkenntnisse effektiver in praxisnahe Lösungen umsetzen wird.
Referenzlinks:
https://mp.weixin.qq.com/s/usrVEOeBD5gphhslZahLCA
https://mp.weixin.qq.com/s/Pxlh69TXSr8ffAp_ul3URw








