Command Palette
Search for a command to run...
Ein Team Der Chinesischen Universität Hongkong, Der Zhejiang-Universität Und Der Polytechnischen Universität Macau Hat Ein Allgemeines Rahmenwerk, Bi-TEAM, Vorgeschlagen, Um Die Genauigkeit Der Vorhersage Hämolytischer Erkrankungen Durch 350% Zu Verbessern, Indem Es Biologische Semantik Und Chemische Präzision integriert.

In der Biochemie und im Molekulartechnik entwickelt sich das Charakterisierungslernen zunehmend zu einer Schlüsseltechnologie, um molekulare Funktionen aufzuklären und die Entwicklung therapeutischer Moleküle voranzutreiben. Die Qualität der eingebetteten Merkmale bestimmt oft die Leistungsfähigkeit nachfolgender Aufgaben wie der Vorhersage von Peptideigenschaften und dem De-novo-Design. Als zentrales Molekül, das biologische Funktionen und chemische Eigenschaften verbindet, ist die Modellierung von Peptidstruktur und -funktion von großer Bedeutung für die Arzneimittelentwicklung.In den letzten Jahren hat die Einführung nicht-klassischer Aminosäuren den Funktionsraum von Peptiden erheblich erweitert und deren Stabilität und Bioverfügbarkeit verbessert, aber komplexe chemische Modifikationen haben auch neue Herausforderungen für traditionelle Modellierungsmethoden mit sich gebracht.Die Frage, wie sich biologische Evolutionsinformationen und chemische Rationalität gleichzeitig in ein Modell integrieren lassen, entwickelt sich zu einem Schlüsselthema, das in diesem Bereich dringend angegangen werden muss.
Aktuell wird die Peptidmodellierung hauptsächlich über zwei technische Wege durchgeführt.einerseits,Protein-Sprachmodelle wie ESM und ProtT5 erfassen den biologischen Kontext und evolutionäre Informationen durch umfangreiches Sequenz-Vortraining und liefern so übertragbare biologische Repräsentationen für nachfolgende Aufgaben.auf der anderen Seite,Um das Problem der Modifikationen nicht-klassischer Aminosäuren anzugehen, verwendeten die Forscher ein chemisches Sprachmodell, um chemische Details durch Wortsegmentierung auf atomarer Ebene zu erfassen und so die Mängel von Proteinmodellen auf chemischer Ebene auszugleichen.
Beide Modelltypen weisen jedoch inhärente Einschränkungen auf. Proteinsprachmodelle sind durch den natürlichen Zeichensatz der Aminosäuren begrenzt, was die Behandlung nicht-klassischer Reste erschwert. Bestehende Methoden zur Annäherung oder Erweiterung des Vokabulars führen häufig zu Verzerrungen oder semantischer Sparsamkeit. Chemische Sprachmodelle hingegen ignorieren den globalen biologischen Kontext, und eine dichte Wortsegmentierung überschreitet leicht das Kontextfenster, was die Anpassung an die Modellierung langer Sequenzen erschwert. Auch allgemeine Modelle leiden unter Domänenverzerrungen.
Um die zuvor genannten Probleme anzugehen, hat die Chinesische Universität Hongkong in Zusammenarbeit mit der Polytechnischen Universität Macau, der Universität Zhejiang, dem Xiangya Second Hospital der Central South University und der Universität für Elektronische Wissenschaft und Technologie Chinas ein selektives Fusionsmodellierungsparadigma vorgeschlagen.Ausgehend von dem Verständnis, dass „chemische Variation eine lokale Störung des biologischen semantischen Raums darstellt“, wurde ein allgemeines Rahmenwerk namens Bi-TEAM entwickelt, um lokale chemische Variationen in den globalen Proteinhintergrund einzufügen.
Dieses Framework nutzt biologische Repräsentationen als semantische Grundlage und erreicht durch die adaptive Einbindung chemischer Signale eine effektive Fusion von biologischen Evolutionsinformationen und chemischen Erkenntnissen. In verschiedenen Aufgaben übertrifft Bi-TEAM durchweg die modernsten Basismodelle: Bei einer strengen Datenpartitionierung basierend auf Skelettähnlichkeit verbessert sich der Matthews-Korrelationskoeffizient um bis zu 661 TP³T; bei der Hämolysevorhersage wird die Genauigkeit um 3501 TP³T gesteigert.
Die zugehörigen Forschungsergebnisse mit dem Titel „Bi-TEAM: Ein einheitliches, skalenübergreifendes Repräsentationslernframework für chemisch modifizierte Biomoleküle“ wurden als Preprint auf arXiv veröffentlicht.
Forschungshighlights:
Das Bi-TEAM-Framework kann multiskalige biochemische Eigenschaften adaptiv integrieren und als hochpräzises Vormodell für ein effizientes Peptiddesign dienen.
* Die Forscher evaluierten Bi-TEAM umfassend anhand von 10 verschiedenen Datensätzen aus 3 biochemischen Domänen und erzielten dabei eine Leistung auf dem neuesten Stand der Technik (SOTA) bei 7 wichtigen Vorhersageaufgaben.
* Dieses Modell erzielt einen doppelten Durchbruch bei Vorhersage- und Generierungsaufgaben, indem es die MCC um 66% unter strenger Segmentierung der Gerüstähnlichkeit verbessert und gleichzeitig die Erfolgsrate beim Entwurf zellpenetrierender cyclischer Peptide um fast das Vierfache erhöht.

Papieradresse:
https://arxiv.org/abs/2603.01873
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „Bi-TEAM“, um das vollständige PDF zu erhalten.
Es wurde eine umfassende Auswertung durchgeführt, die drei wichtige biochemische Bereiche und zehn verschiedene Datensätze umfasste.
Diese Studie bewertet die Eigenschaften aus zwei Perspektiven: Vorhersage der Eigenschaften und gezielte Generierung. Sie umfasst drei wichtige Forschungsbereiche: modifizierte Peptide, posttranslationale Modifikationen (PTMs) und natürliche Proteine und beinhaltet insgesamt 10 Datensätze.
Im Bereich der modifizierten Peptide konzentriert sich die Forschung auf die Bewertung der Fähigkeit des Modells, die Membranpermeabilität vorherzusagen.Die Kerntrainingsdaten stammen aus der ProPAMPA-Datenbank, die 12–46 Ringatome enthält und deren Sequenzlängenverteilung annähernd normal ist, jedoch an beiden Enden signifikante lange Ausläufer aufweist. Sie enthält zudem eine große Anzahl natürlicher und nicht-klassischer Aminosäurereste und belegt damit eine hohe chemische Diversität. Nach der Deduplizierung mit RDKit,Es enthält insgesamt 6.876 nicht-konjugierte cyclische Peptidsequenzen.
Zur Bewertung der Generalisierungsfähigkeit des Modells wurden drei externe Datensätze aus Nassversuchen herangezogen: ProCacoPAMPA, CycPeptMPDB v1.2 und der Rezai-Datensatz. Diese Datensätze umfassen cyclische Peptidproben unterschiedlicher Länge und Struktur. Im Einzelnen:
ProCacoPAMPA:Alle transmembranären cyclischen Peptidsequenzen der Längen 6 und 10 wurden aus bestehenden Studien zusammengetragen und zu einem standardisierten Datensatz zusammengestellt.
CycPeptMPDB v1.2:Die neueste Version der größten öffentlich zugänglichen Datenbank zur Membranpermeabilität nicht-klassischer cyclischer Peptide, die aus 56 Publikationen zusammengestellt wurde, umfasst 8.466 Datensätze. In dieser Studie entfernten die Forscher Duplikate aus dem ProPAMPA-Datensatz und erhielten so einen verfeinerten Datensatz mit 1.230 Datenpunkten.
Rezai:Dies umfasst Daten zur passiven Membranpermeabilität für 11 cyclische Peptide. Diese Daten wurden mittels PAMPA-Experimenten gewonnen und werden häufig zur externen Modellvalidierung unter Bedingungen mit kleinen Proben verwendet.
Um die Arzneimittelähnlichkeit und den Krankheitszusammenhang des Modells weiter zu bestätigen,Die Forscher führten eine Vorhersageaufgabe zur Arzneimittelähnlichkeit anhand des PTM-Datensatzes durch.Die verwendeten Daten umfassten zwei Kategorien: Datensätze zu Arzneimitteln und krankheitsassoziierte Datensätze. Erstere bestanden hauptsächlich aus längeren Proteinsequenzen, deren Modifikationsstellen eine ausgeprägte Verteilung mit langem Ausläufer aufwiesen. Letztere stammten primär aus Datenbanken wie dbPTM und genomweiten Assoziationsstudien (GWAS). Obwohl die Verteilung der Modifikationsstellen ähnlich war wie bei den ersteren, war der Bereich der Sequenzlängen größer, wodurch ein vielfältigerer struktureller Kontext entstand.
Im Bereich der natürlichen Proteine konzentrierten sich die Forscher auf die Bewertung der Leistungsfähigkeit des Modells bei der Vorhersage von Löslichkeit und Hämolyse, um die Schlüsselmechanismen der Peptidhämolyse und Veränderungen der Proteinlöslichkeit zu erforschen. Die verwendeten Datensätze umfassten hauptsächlich drei Kategorien: Hämolyse, Antikontamination und Löslichkeit.
Die Hämolysedaten stammen aus der DBAASP v3-Datenbank.Es enthält insgesamt 9.316 Sequenzen, die aus klassischen Aminosäuren vom L-Typ bestehen.
Der Anti-Umweltverschmutzungs-Datensatz besteht hauptsächlich aus kurzen Peptidsequenzen.Die Kettenlängen konzentrieren sich auf 5 bis 10 Aminosäurereste, und ihre LogP-Verteilung ist annähernd normalverteilt. Die Proben weisen eine gute Clusterstruktur im Merkmalsraum auf.
Der Datensatz zur Löslichkeit basiert auf Proteinsequenzen, die mit PROSO II annotiert wurden.Die Bezeichnung basiert auf einer retrospektiven Analyse der Protein Structure Initiative.
Bi-TEAM: Ein einheitliches, skalenübergreifendes Charakterisierungslernframework für chemisch modifizierte Biomoleküle
Bi-TEAM zielt darauf ab, die Herausforderung bestehender unimodaler Modelle zu bewältigen, indem es gleichzeitig globale evolutionär-biologische Informationen und lokale chemische Strukturdetails (feinkörniger chemischer Raum) erfasst. Wie in der folgenden Abbildung dargestellt,Die Kernidee besteht darin, ein dual-perspektivisches Repräsentationssystem zu konstruieren, das den evolutionär-biologischen Raum und den chemischen Strukturraum tiefgreifend integriert.Dies ermöglicht genauere Modellierungsfähigkeiten für Peptidsequenzen, die nicht-klassische Aminosäuren enthalten.
In Bezug auf die Gesamtarchitektur,Das Modell nutzt den vom Protein-Sprachmodell konstruierten biologischen Raum als semantisches Rückgrat und schöpft dabei die aus groß angelegten natürlichen Sequenzen gelernten evolutionären Muster und Kontextbeziehungen voll aus.Parallel dazu wird ein chemisches Sprachmodell (CLM) eingeführt, um Strukturinformationen auf atomarer Ebene zu erfassen und so die inhärenten Einschränkungen des Protein-Sprachmodells (PLM) bei der Verarbeitung chemischer Modifikationen auszugleichen. Die beiden Modelltypen ergänzen sich auf der Repräsentationsebene und erweitern gemeinsam die Ausdruckskraft der Eingabesequenz.

Bei der Verarbeitung modifizierter Peptidsequenzen kodiert Bi-TEAM diese mithilfe zweier komplementärer Informationsströme:Eine davon ist ein biologischer Sequenzstrom.Modifizierte Aminosäuren werden den strukturell ähnlichsten natürlichen Aminosäuren zugeordnet. Dadurch wird eine Erweiterung der Wortsegmentierungstabelle vermieden und die evolutionäre Semantik, die für die Modellierung genutzt werden kann, bleibt erhalten.Die andere ist die SELFIES-ähnliche Darstellungsform.Es dient dazu, die Veränderungen der funktionellen Gruppen und die chemischen Bindungsstrukturen modifizierter Reste auf atomarer Ebene genau zu beschreiben und so stabile Strukturinformationen für chemische Sprachmodelle bereitzustellen.
Nach Abschluss der Dual-Stream-CodierungDas Modell wird mithilfe eines dual-gated residualen Mechanismus fusioniert, der durch positionsabhängige Verzierungshinweise gesteuert wird:Unter Verwendung biologischer Repräsentationen als semantische Grundlage werden wichtige chemische Signale mithilfe von Gating-Einheiten gefiltert und eingefügt, wobei verbleibende Verbindungen biologischer Merkmale erhalten bleiben. Dies ermöglicht dem Modell, eine effektive Korrelation zwischen globalen Sequenzbeschränkungen und lokalen chemischen Veränderungen herzustellen und gleichzeitig die Stabilität des Trainings zu gewährleisten.
Auf Anwendungsebene zeichnet sich Bi-TEAM durch eine gute Vielseitigkeit aus.Bei der Verarbeitung unveränderter natürlicher Proteinsequenzen kann das Modell die Mapping- und Lokalisierungsschritte direkt überspringen und sich so an routinemäßige Proteinaufgaben anpassen, ohne die Gesamtarchitektur zu verändern.
Was Trainingsstrategien angeht,Die Studie verwendet ein zweistufiges Rahmenkonzept aus „Vortraining und Feinabstimmung“:Zunächst wurde ein domänenadaptives Vortraining zweier Arten von Basis-Encodern auf Basis natürlicher Proteinsequenzen bzw. Korpora chemischer Verbindungen kleiner Moleküle durchgeführt. Anschließend lernte das Modell durch gemeinsames Multi-Task-Feintuning die Fusionsregeln biologischer und chemischer Merkmale in verschiedenen Aufgabenszenarien und verbesserte so seine Generalisierungsfähigkeit weiter.
Bi-TEAM hat einen Durchbruch bei der Entwicklung penetrierender cyclischer Peptide erzielt und die Erfolgsquote um das 4,6-fache erhöht.
Um die Anwendbarkeit von Bi-TEAM in unbekannten chemischen Bereichen zu überprüfen, konzentriert sich diese Studie auf das Design nicht-kanonischer cyclischer Peptide für die zellpenetrierende Behandlung der neovaskulären altersbedingten Makuladegeneration (nAMD) mittels nicht-invasiver Wirkstoffverabreichung. Die Studie führte systematisch ein vollständiges Prozess-Experiment mit „vorhersagegestützter Analyse“ durch, um die Leistungsfähigkeit des Modells im eigenschaftsbasierten Moleküldesign zu evaluieren.
Die neovaskuläre altersbedingte Makuladegeneration (nAMD) ist eine Hauptursache für irreversible Erblindung im Alter. Ihre zentrale Pathologie besteht in der VEGF-vermittelten choroidalen Neovaskularisation und Permeabilitätsstörung. Die klinische Behandlung beruht derzeit hauptsächlich auf der intravitrealen Injektion von großmolekularen Anti-VEGF-Medikamenten (wie z. B. Aflibercept, 115 kDa).Allerdings haben diese Medikamente Schwierigkeiten, die physiologische Barriere des Auges zu durchdringen, und langfristige Injektionen können zu Komplikationen und Compliance-Problemen führen.Die Entwicklung von Peptidbindern, die spezifisch an Aflibercept binden und dessen Transport durch die Blut-Hirn-Schranke fördern, eröffnet neue Möglichkeiten für die nicht-invasive Augentropfentherapie. Im Vergleich zu leicht abbaubaren linearen Peptiden mit kurzer Halbwertszeit gelten cyclische Peptide aufgrund ihrer stabileren Struktur und höheren Permeabilität als idealere Wirkstoffträger. Dies ist der Hauptgrund für das Interesse der Forschung an der Entwicklung cyclischer Peptide.
In dieser Studie wurde zunächst eine Vorhersage und Bewertung von zellpenetrierenden Peptiden (CPPs) durchgeführt, um eine Grundlage für nachfolgende Generierungsaufgaben zu schaffen.Der Datensatz wurde gemäß dem pLM4CPP-Standardschema erstellt und integrierte Datenbanken wie CPPsite2.0, C2Pred und CellPPD. Nach Screening und Deduplizierung wurden 1.399 positive Proben (experimentell validierte penetrierende Peptide) und 4.080 negative Proben erhalten. Zu den Vergleichsmodellen gehörten gängige Protein-Embedding-Modelle wie SeqVec, ESM2 und ProtT5. Die Bewertungsmetriken umfassten ACC, BACC, Sn, Sp, MCC und AUC.
Die Ergebnisse zeigen, dass Bi-TEAM in allen Bereichen die beste Leistung erzielt hat:Die Genauigkeit (ACC) verbesserte sich um 5,521 TP3T im Vergleich zu SeqVec, die absolute Genauigkeit (BACC) um 5,881 TP3T im Vergleich zu ESM2-480, die Sensitivität (Sn) um 12,581 TP3T, die Spezifität (Sp) um 1,451 TP3T im Vergleich zu ProtT5-XL BFD, der Matthews-Korrelationskoeffizient (MCC) um 14,681 TP3T im Vergleich zu SeqVec und die Fläche unter der Kurve (AUC) um 8,451 TP3T im Vergleich zu ESM2-480. Die signifikanten Verbesserungen der Sensitivität und des MCC zeigen, dass das Modell einen deutlichen Vorteil bei der Identifizierung von penetrierenden Peptiden aufweist.
Auf dieser Grundlage werden weitere Forschungen zu eigenschaftsgesteuerten cyclischen Peptidgenerierungsexperimenten durchgeführt.Unter Verwendung von BoltzDesign1 als Basisframework wurden 1.000 cyclische Peptide der Länge 10-20 unter zwei Bedingungen generiert: einer unter Verwendung nur der standardmäßigen Strukturbeschränkungen und der anderen unter Einführung von Bi-TEAM als zusätzlichem Gradientenleitfaden während des Generierungsprozesses.
Das Erfolgskriterium war ein Bi-TEAM-Vorhersage-Log-Odds-Wert größer als 0,5. Die Ergebnisse zeigten, dass…Bei herkömmlichen Methoden wurde eine Erfolgsquote von lediglich 6,71 TP3T bei der Herstellung zellpenetrierender cyclischer Peptide erzielt, während Bi-TEAM-gestützte Methoden diese auf 30,71 TP3T verbesserten.Gleichzeitig verschlechterte sich die Strukturqualität nicht: Der durchschnittliche pLDDT-Wert des generierten Peptid-Aflibercept-Komplexes lag über 0,82, was darauf hindeutet, dass das Modell eine gute strukturelle Zuverlässigkeit und Stabilität der Bindungsschnittstelle beibehielt und gleichzeitig die Penetration verbesserte.

Um den Steuerungsmechanismus zu verstehen, analysierten die Forscher die Restmuster der generierten Sequenz. Frühere Studien haben gezeigt, dass das hydrophobe Triplett aus Tryptophan (W), Phenylalanin (F) und Tyrosin (Y) sowie positiv geladene Reste wie Arginin (R) und Lysin (K) Schlüsseleigenschaften für zellpenetrierende Peptide sind, um den Membrantransport zu ermöglichen.
Die Analyse ergab, dassMithilfe von Bi-TEAM konnte die Häufigkeit des gemeinsamen Auftretens von hydrophoben Tripletts und zwei positiv geladenen Resten in der generierten Sequenz signifikant erhöht werden, und auch die Verteilung der Restanzahl zeigte einen konsistenten Trend.Dieses Anreicherungsmuster stimmt weitgehend mit den bekannten Struktur-Funktions-Beziehungen membrangängiger Peptide überein. Dies deutet darauf hin, dass Bi-TEAM nicht nur relevante biologische Mechanismen erfassen, sondern auch die Wahrscheinlichkeit für Sequenzen mit membrangängigen Eigenschaften während des Generierungsprozesses signifikant erhöhen kann. Die Analyse der Kontrollvariablen schloss zudem den Einfluss der Peptidlänge (10–20 Aminosäuren) aus und zeigte damit, dass das Modell die Verteilung der Sequenzen tatsächlich in einen für den Membrantransport günstigeren chemo-biologischen Bereich lenkt.
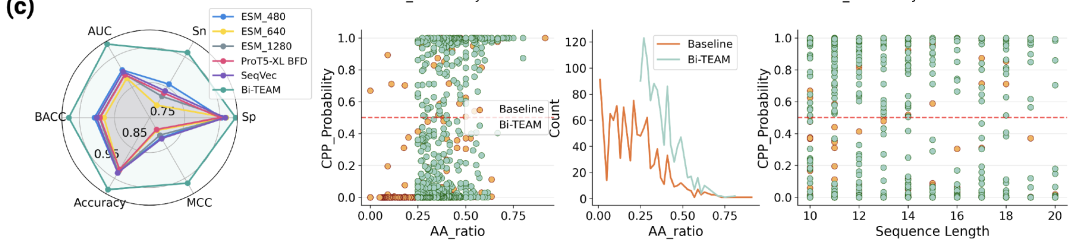
Im mittleren Abschnitt: die Beziehung zwischen der Häufigkeit wichtiger hydrophober Reste und der Penetrationswahrscheinlichkeit;
Rechts: Zusammenhang zwischen der Länge des zyklischen Peptids und der Penetrationswahrscheinlichkeit
Abschließend validierte die Studie die Ergebnisse auf struktureller Ebene anhand einer Fallstudie. Die Forscher visualisierten zunächst die dreidimensionale Struktur des Aflibercept-Dimers und färbten dessen Moleküloberfläche basierend auf dem elektrostatischen Potenzial ein. Anschließend nutzten sie AlphaFold3, um die Komplexstruktur des cyclischen Peptids mit Aflibercept vorherzusagen und zu designen. Die Analyse identifizierte zwei potenzielle Bindungstaschen für das cyclische Peptid: eine hydrophobe Kavität aus drei Ringen und eine weitere, die aus einer Ringstruktur und einem β-Faltblattfragment besteht. Diese Strukturinformationen bilden eine wichtige Grundlage für die weitere Optimierung des cyclischen Peptids und potenzielle klinische Anwendungen.

Schwerpunkt auf technologischer Innovation im Bereich der Peptid-Arzneimittelentwicklung
Auf dem Gebiet der Peptidwissenschaft erforschen zahlreiche Forschungseinrichtungen weltweit aktiv neue technologische Wege und Behandlungsmöglichkeiten, um schwere Krankheiten zu besiegen – von der Grundlagenforschung bis zur klinischen Anwendung.
Zum Beispiel das Team für Strukturbiologie an der School of Biochemistry der Universität Bristol, Großbritannien.Fortgeschrittene Techniken wie die Kryo-Elektronenmikroskopie und die Röntgenkristallographie werden zur Analyse der Feinstruktur des Immunsystems eingesetzt, und auf dieser Grundlage wird ein strukturgeleitetes Peptidwirkstoffdesign durchgeführt.Sie versuchen, Wirkstoffkandidaten der nächsten Generation für die Behandlung von Autoimmunerkrankungen zu entwickeln, indem sie zyklische Peptidmoleküle entwerfen, die das menschliche Komplementsystem präzise aktivieren können.
Das ToxiCode-Projekt, eine Kooperation zwischen dem King's College London und der Universität Zagreb, erforscht derweil einen einzigartigen Weg zur Entdeckung neuer Medikamente aus Tiergiften.Das Projekt kombiniert künstliche Intelligenz und synthetische Biologie und nutzt ein hybrides KI-System, um Peptidsequenzmuster und deren Struktur-Aktivitäts-Beziehungen zu erlernen. Dies ermöglicht die schnelle Entwicklung neuartiger bioaktiver Peptide zur Behandlung von Krebs, neurologischen Störungen und Infektionskrankheiten.Dies bietet einen neuen methodischen Rahmen für eine nachhaltige und ethische Arzneimittelforschung.
Dies zeigt, dass die Entwicklung von Peptidwirkstoffen ein neues Forschungsparadigma prägt: Strukturbiologie, künstliche Intelligenz und chemische Biologie konvergieren zunehmend, und die Grenzen zwischen Grundlagenforschung und industrieller Entwicklung verschwimmen immer mehr. Neue Moleküle entstehen oft aus interdisziplinären Technologiekombinationen, doch entscheidend für ihren klinischen Einsatz ist der sich stetig etablierende Translationsweg von der Entdeckung im Labor zur industriellen Anwendung. In diesem Prozess werden Peptidmoleküle aufgrund ihrer einzigartigen Eigenschaften, die zwischen kleinen und großen Molekülen liegen, neu bewertet und finden in immer mehr Krankheitsbereichen Anwendung.
Referenzlinks:
1.https://www.bristol.ac.uk/news/2025/november/bristol-researcher-awarded-over-850000-to-develop-new-treatments.html
2.https://www.kcl.ac.uk/news/kings-to-collaborate-in-venom-based-drug-discovery-project
3.https://mp.weixin.qq.com/s/X67D1qrUzclwOsJ9cKUtZg








