Command Palette
Search for a command to run...
Das MIT Hat VibeGen Vorgeschlagen, Das Erste Durchgängige Dynamische Proteingenerierungsmodell, Das Eine Bidirektionale Abbildung Zwischen Sequenz Und Vibration ermöglicht.
Proteine sind die zentralen Funktionsmoleküle lebender Systeme, doch ihre Funktionen werden nicht allein durch ihre statische Struktur bestimmt, sondern resultieren aus ihrer sich ständig verändernden Konformationsdynamik. Innerhalb eines komplexen Energieumfelds halten Proteine unter physiologischen Bedingungen durch Bewegungen auf verschiedenen Skalen – von Femtosekunden bis Millisekunden – ein dynamisches Gleichgewicht aufrecht und sind damit wahre molekulare Maschinen.
Aus diesem Grund stehen abnorme Proteindynamiken in engem Zusammenhang mit einer Vielzahl von Erkrankungen. Beispielsweise ist die Funktion des Tumorsuppressorproteins p53 von der Konformationsplastizität abhängig, und onkogene Mutationen schwächen diese Fähigkeit; CFTR-Mutationen hingegen induzieren Mukoviszidose, indem sie die Kanalöffnungsdynamik stören. Diese Tatsachen deuten darauf hin, dass…Die „Bewegung“ von Proteinen ist selbst ein wichtiger Faktor für deren Funktion.Daher wird das Verständnis und die Gestaltung von Proteinen aus einer dynamischen Perspektive zu einer zukunftsweisenden Richtung in der Strukturbiologie und im Bioengineering.
In den letzten Jahrzehnten haben Forscher experimentelle Techniken wie die Kernspinresonanz (NMR), die Wasserstoff-Deuterium-Austausch-Massenspektrometrie (HDEMS) und die Kryo-Elektronenmikroskopie (Kryo-EM) sowie computergestützte Methoden wie Molekulardynamiksimulationen und die Analyse von Schwingungsmoden (VMS) entwickelt, um die Proteindynamik zu charakterisieren. Diese Methoden sind jedoch entweder zu komplex für eine groß angelegte Anwendung oder zu rechenaufwändig und zeitintensiv, wodurch sie für groß angelegte Studien ungeeignet sind.
In den letzten Jahren haben Deep Learning und generative KI neue Möglichkeiten für die Proteinforschung eröffnet. Modelle wie AlphaFold2 ermöglichen hochpräzise Strukturvorhersagen, und die Methoden können auch Sekundärstrukturen, Bindungsstellen und sogar Schwingungsmerkmale vorhersagen.Die meisten bestehenden Methoden verharren noch auf der Ebene der „Struktur oder einzelner Eigenschaften“ und vernachlässigen die systematische Modellierung der intrinsischen Dynamik.Im Bereich des Designs behandeln Frameworks wie RFdiffusion und AlphaFold3 Strukturen weiterhin als approximative starre Körper und haben dynamische Randbedingungen noch nicht vollständig integriert. Daher bleibt die Etablierung einer einheitlichen Abbildung von „Sequenz-Struktur-Dynamik-Funktion“ und die Realisierung eines kontrollierbaren Designs auf Basis von Dynamik eine zentrale Herausforderung.
Kürzlich,Ein gemeinsames Forschungsteam des MIT und der Carnegie Mellon University hat VibeGen entwickelt, einen intelligenten Agenten, der Proteine erzeugt.Durch die Kombination von Sequenzgenerierung und Schwingungsdynamikvorhersage wurde ein neuartiges Proteindesign realisiert. Die Ergebnisse zeigen, dass die mit diesem generativen Verfahren entworfenen Proteine nicht nur stabile und neuartige Strukturen bilden können, sondern auch die Verteilungseigenschaften der Zielschwingungsamplituden auf der Hauptkettenebene reproduzieren.
Die zugehörigen Forschungsergebnisse mit dem Titel „VibeGen: Agentic end-to-end de novo protein design for tailor dynamics using a language diffusion model“ wurden in Matter veröffentlicht.
Papieradresse:
https://www.cell.com/matter/abstract/S2590-2385(26)00069-X
Proteindynamikdatenbank basierend auf niederfrequenten Normalschwingungsmoden
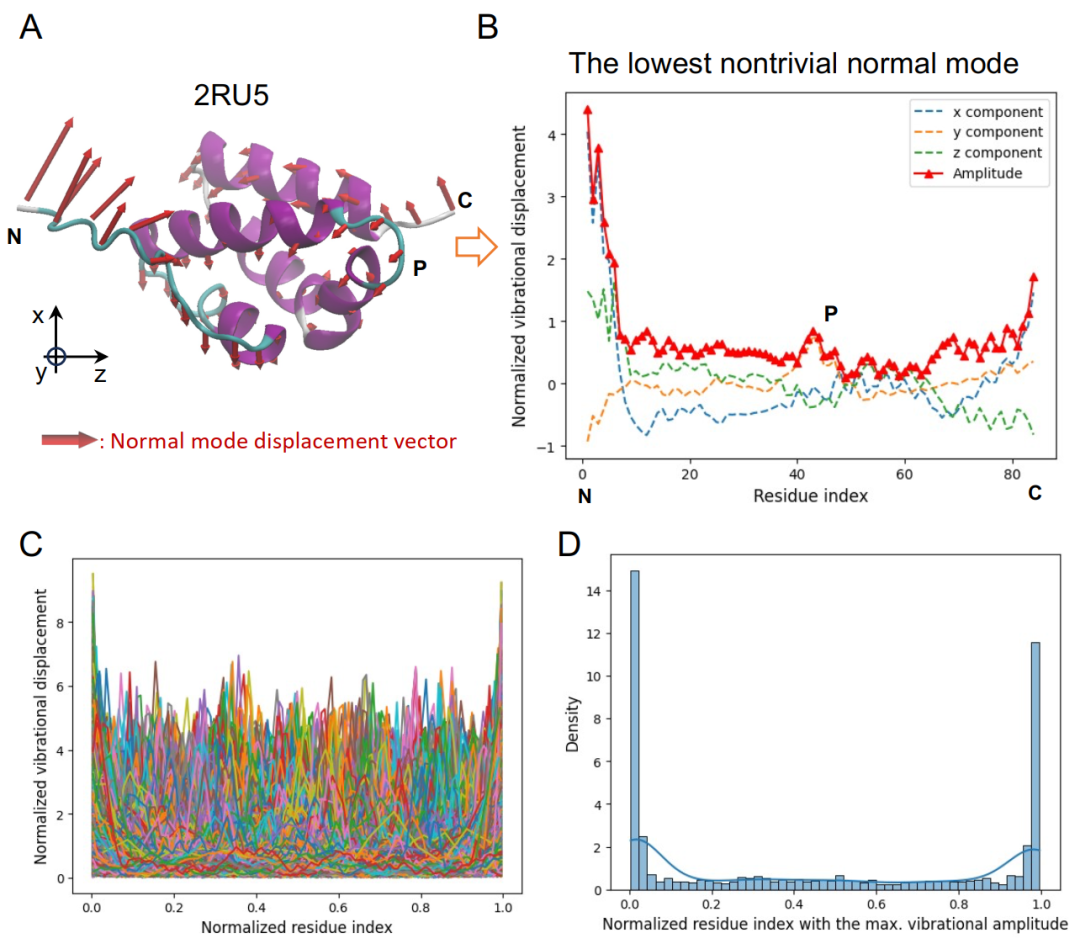
Zum Aufbau der DatenbankForscher durchsuchten die im Januar 2024 aktualisierte Proteindatenbank (PDB) nach einkettigen Proteinen mit einer Länge von höchstens 126 Aminosäuren.Die Struktur wurde mithilfe von Tools wie VMD, MMTSB und SCWRL4 bereinigt und vervollständigt. Anschließend wurde eine Energieminimierung auf Basis des CHARMM-Kraftfelds durchgeführt und die Modalinformationen mithilfe der Blocknormalschwingungsmethode berechnet. Nach dem Entfernen der ersten sechs Starrkörpermoden, die die Gesamttranslation und -rotation repräsentieren, wurde die niedrigste nichttriviale Mode für die weitere Analyse ausgewählt.
Darauf aufbauend extrahierte die Studie die Schwingungsmoden der Cα-Atome an jedem Rest der Hauptkette und erstellte einen Vektor der normalen Schwingungsmoden. Die Ergebnisse zeigten eine deutlich heterogene Verteilung der Schwingungsamplituden: größere Amplituden an den Kettenenden und in locker strukturierten Bereichen, während die Schwingungen in dichten Bereichen wie α-Helices und β-Faltblättern eingeschränkt waren. Schleifen- und Knäuelbereiche wiesen aufgrund ihrer höheren Flexibilität lokale Maxima auf. Um den Einfluss von Längenunterschieden zu eliminieren, wurden die Vektoren normalisiert, wodurch sie zu koordinatenunabhängigen dynamischen Deskriptoren wurden.
Finale,Die Forscher erstellten einen Datensatz mit 12.924 Protein-Einzelsträngen.Die Analyse zeigt, dass die niederfrequenten Schwingungsmoden sehr vielfältig sind, wobei die Amplitudenspitzen an den Kettenenden konzentriert sind. Der Datensatz wurde im Verhältnis 9:1 in Trainings- und Testdatensätze aufgeteilt, um das generative Modell anschließend zu trainieren und zu evaluieren.
VibeGen: End-to-End-De-novo-Protein-Design basierend auf einem Sprachdiffusionsmodell
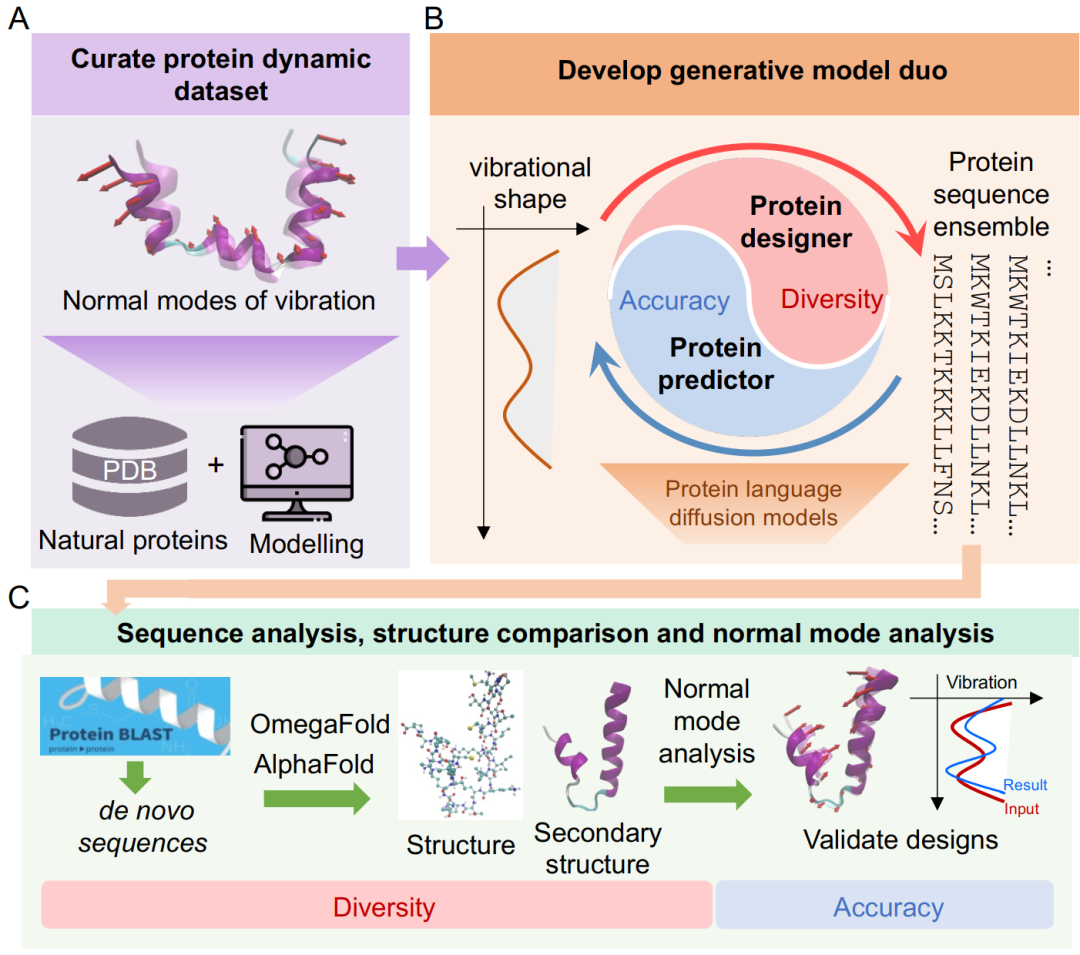
Die zentrale Herausforderung dieser Studie besteht darin, dass die Form der normalen Schwingungsmoden durch die komplexe dreidimensionale Struktur und die elastischen Eigenschaften von Proteinen bestimmt wird und keine direkte Beziehung zwischen Sequenz und Dynamik besteht. Gleichzeitig sind Einzelmodeninformationen stark degeneriert, und unterschiedliche Sequenzen können ähnlichen dynamischen Eigenschaften entsprechen, was das inverse Designproblem besonders schwierig macht.
Um diese Herausforderungen zu bewältigen, wurden in dieser Studie zunächst wichtige dynamische Merkmale einer großen Anzahl von Proteinen aus einer Proteindatenbank (PDB) mittels Normalschwingungsanalyse und Allatom-Molekulardynamiksimulationen extrahiert. Darauf aufbauendDie Forscher konstruierten zwei kollaborative Protein-Sprachdiffusionsmodelle: ein Protein-Designmodul (PD) und ein Vorhersagemodul (PP).Sie sind für die Vorwärtsvorhersage bzw. die inverse Konstruktion zwischen der Sequenz und dem normalen Schwingungsmodenraum verantwortlich. Die beiden Module weisen ähnliche Strukturen auf und basieren beide auf einer Kombination aus einem vortrainierten Protein-Sprachmodell (pLM) und einem Diffusionsmodell.
Die Aufgabe des Designmoduls besteht darin, eine Sequenz auf Basis der angestrebten dynamischen Eigenschaften zu generieren.Während des Entrauschungsprozesses integriert das Diffusionsmodell dynamische Zustandsinformationen über mehrere Kanäle und erzeugt schrittweise Sequenzen, die den Zielcharakteristika im latenten Raum entsprechen.Das Vorhersagemodul besitzt eine symmetrische Struktur und leitet die Form der normalen Schwingungsform aus der Eingangssequenz ab. Es nutzt außerdem mehrere Sequenzdarstellungen, die vom vortrainierten Sprachmodell ausgegeben werden, um die Vorhersageergebnisse zu optimieren.
Die beiden Module werden unabhängig voneinander trainiert und bilden zusammen während der Einsatzphase ein geschlossenes, kollaboratives System aus „Generierung-Evaluierung-Screening“.Das Designmodul generiert zunächst Kandidatensequenzen, und das Vorhersagemodul bewertet deren dynamische Leistung in Echtzeit.Forscher können die Ergebnisse nach ihren Anforderungen an Genauigkeit oder Diversität filtern und die Iterationen so oft wiederholen, bis eine zufriedenstellende Sequenz erreicht ist.
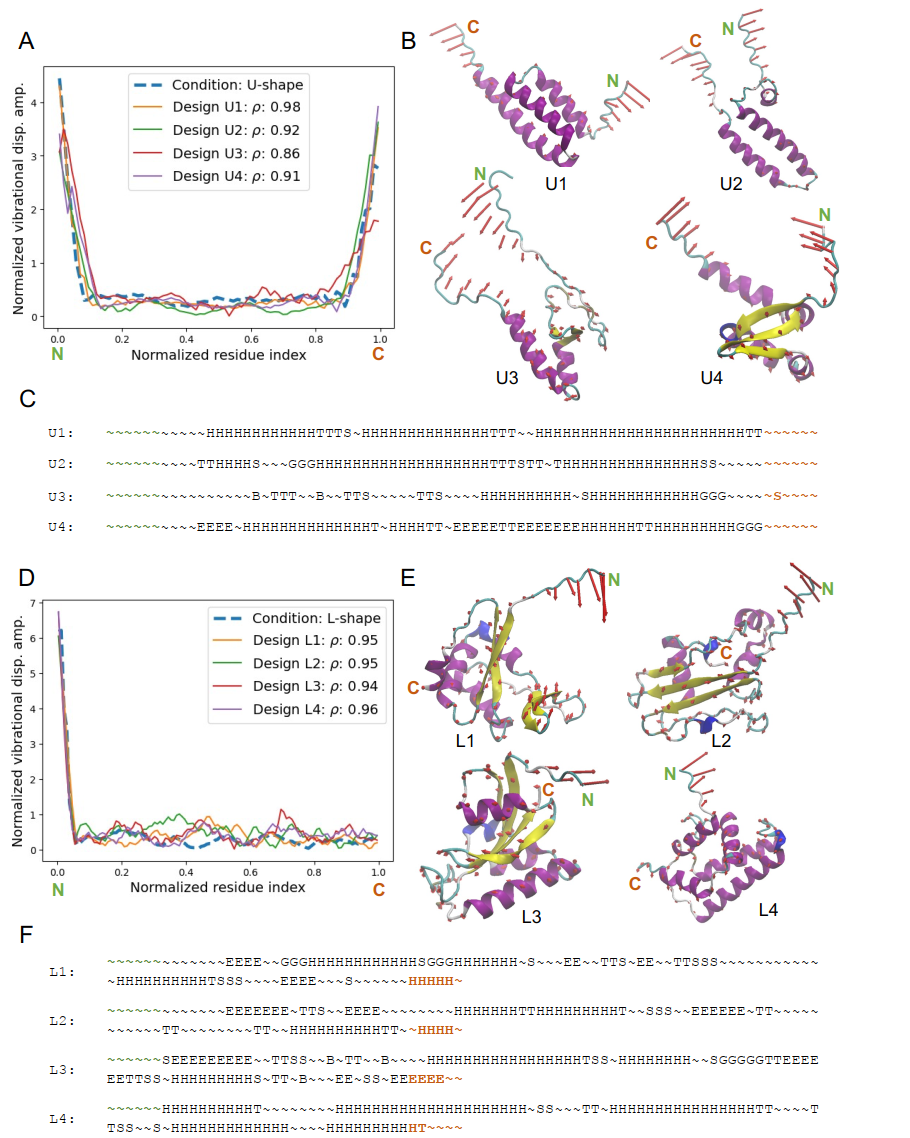
Die Leistungsfähigkeit des Modells wurde anhand des Testdatensatzes validiert. Für verschiedene typische Normalschwingungsformen, darunter L-, U- und W-förmige, wurde das vom Modell generierte Protein durch eine Analyse realer Normalschwingungen validiert, und seine Schwingungsform stimmte sehr gut mit dem Designziel überein. Quantitative Indikatoren wie der Pearson-Korrelationskoeffizient und der relative L²-Fehler zeigten, dass…Mit dieser Methode lässt sich auch unter komplexen dynamischen Randbedingungen eine hochpräzise Konstruktion erreichen.
Aus struktureller Sicht zeigt die Bildung von Proteinen eine klare kinetische Korrespondenz: Bereiche mit stärkeren Schwingungen neigen dazu, ungeordnete Knäuel oder flexible Fragmente zu bilden, während Bereiche mit eingeschränkten Schwingungen dazu neigen, stabile Strukturen wie α-Helices oder β-Faltblätter zu bilden.Dies beweist, dass das Modell die intrinsische Beziehung zwischen Struktur und Dynamik effektiv erfasst hat.
Auf der Ebene der Modellimplementierung verwenden sowohl das Design- als auch das Vorhersagemodul ein mittelgroßes, vortrainiertes Modell mit 150 Millionen Parametern aus der ESM-2-Serie als pLM, um ein ausgewogenes Verhältnis zwischen Recheneffizienz und Modellleistung zu erzielen. Das Diffusionsmodell integriert bedingte Informationen über mehrere Kanäle eines U-förmigen Netzwerks in den Entrauschungsprozess und wird unabhängig mit dem Adam-Optimierer trainiert.
Ein Durchbruch in Präzision und Neuartigkeit
Zur Bewertung der Modellleistung wurden experimentelle Analysen in verschiedenen Dimensionen durchgeführt. Die Diversitätsanalyse ergab, dass…Für dasselbe dynamische Ziel kann das Modell mehrere Entwurfsschemata mit unterschiedlichen Strukturen, aber gleicher Funktion generieren.Anhand von U- und L-förmigen Normalschwingungsmoden lässt sich zeigen, dass alle entworfenen Proteine eine Struktur aus „dichtem Kern und offenen Enden“ aufweisen: Die Enden bilden ungeordnete Knäuelstrukturen, die Bereichen hoher Amplitude entsprechen; der Kern kann auf verschiedene Weise realisiert werden, beispielsweise als α-Helixbündel oder als Hybridstruktur mit helikaler Faltung, die Bereichen niedriger Amplitude entsprechen. Diese Vielfalt resultiert hauptsächlich aus dem Freiheitsgrad bei der Strukturwahl in den Bereichen niedriger Schwingung, und das Modell erfasst und nutzt diese „vielfältigen Lösungsmöglichkeiten“ erfolgreich.
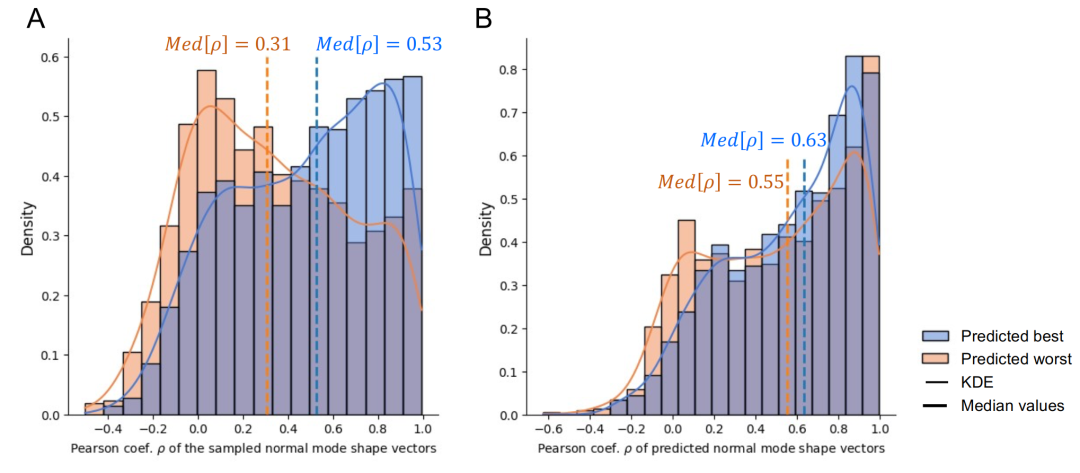
Die Effektivität des Vorhersagemoduls wurde durch Vergleichsexperimente verifiziert. Wie die Abbildung unten zeigt, war die tatsächliche Genauigkeit der Vorhersagen aus den besten und schlechtesten Gruppen desselben Kandidatensequenzsatzes signifikant höher als die der schlechteren (Median des Pearson-Korrelationskoeffizienten: 0,53 vs. 0,31). Das Vorhersagemodul gewährleistete dabei eine stabile Vorhersagegenauigkeit für beide Gruppen. Dies deutet darauf hin, dass…Durch die Einführung eines Vorhersagemoduls während des Entwurfsprozesses können hochwertige Sequenzen effektiv herausgefiltert und die Abhängigkeit von teuren physikalischen Verifikationsverfahren reduziert werden.
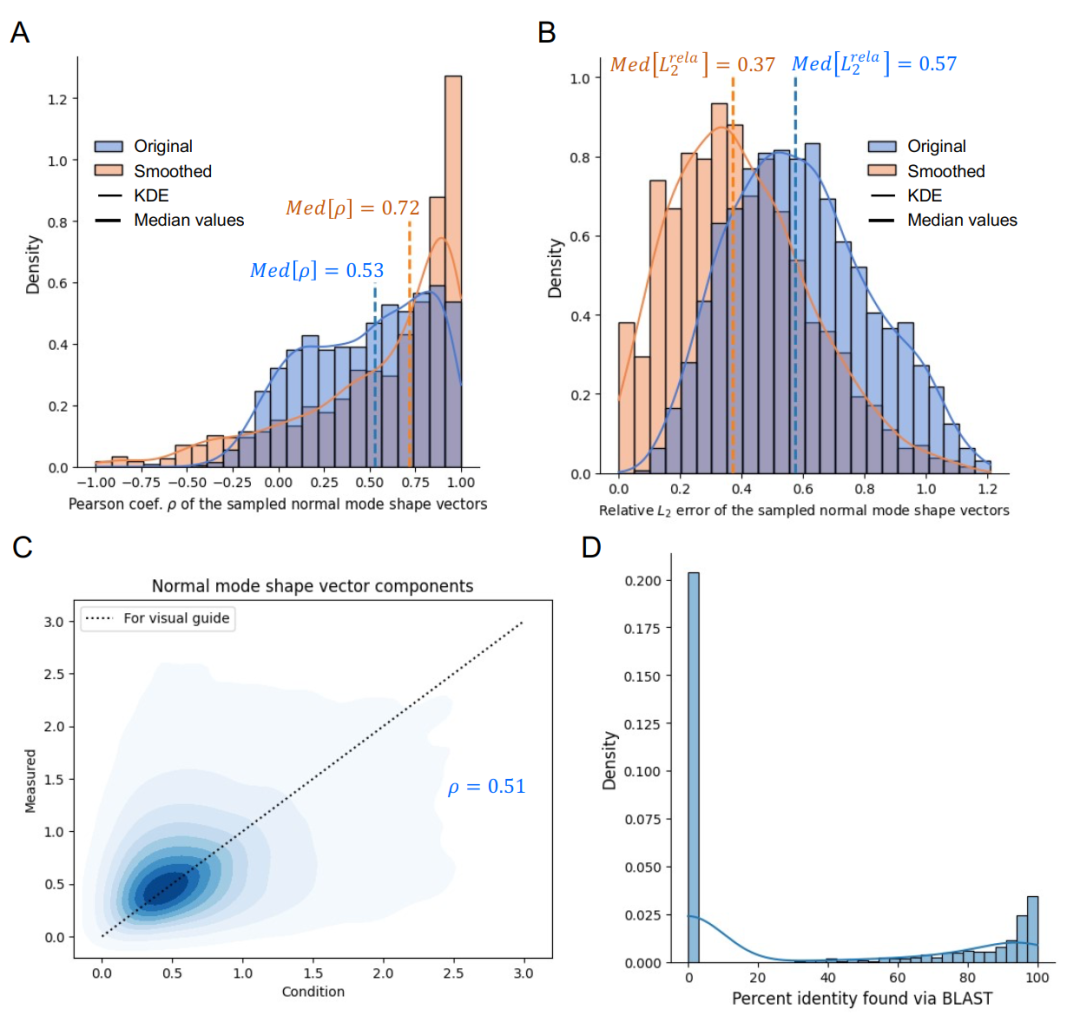
Die Gesamtleistungsstatistik basiert auf 1293 Testfällen. Wie die Abbildung unten zeigt, beträgt der mittlere Korrelationskoeffizient zwischen der gemessenen Eigenschwingungsform und dem Sollwert 0,53, und der mittlere relative L2-Fehler liegt bei 0,57. Dies verdeutlicht die Schwierigkeit, hochpräzise Konstruktionen auf Residuenebene durchzuführen. Nach Anwendung eines Tiefpassfilters zur Erhaltung der Gesamtform steigt der mittlere Korrelationskoeffizient auf 0,72, und der mittlere Fehler sinkt auf 0,37.Dies deutet darauf hin, dass das Modell besonders gut darin ist, das Gesamtprofil der Schwingungen zu erfassen.Dieses Merkmal besitzt die wichtigste biologische Bedeutung für die großräumige Konformationsdynamik von Proteinen.
Hinsichtlich der Neuartigkeit weist die höchste Sequenzidentität von BLAST eine bimodale Verteilung auf, wobei der Hauptgipfel den neu entworfenen Sequenzen entspricht.Dies deutet darauf hin, dass das Modell mit größerer Wahrscheinlichkeit neuartige Sequenzen generiert und somit die potenzielle Bibliothek von Lösungen für Proteinstruktur und -dynamik effektiv erweitert.
Der Zusammenhang zwischen Struktur und Dynamik zeigt sich konsistent in zahlreichen Experimentreihen: Dichte Strukturen wie α-Helices und β-Faltungen sind überwiegend in Bereichen niedriger Amplitude verteilt, während Bereiche hoher Amplitude meist Schleifenbereiche oder Endaufrollungen sind.Das Modell erfasste dieses physikalische Gesetz erfolgreich und war in der Lage, die lokale Flexibilität mithilfe sekundärer Strukturelemente zu steuern, wodurch ein Verständnis des Struktur-Dynamik-Zusammenhangs demonstriert wurde.
Insgesamt erzielt dieses Modell ein gutes Gleichgewicht zwischen Genauigkeit, Vielfalt und Neuartigkeit beim Proteindesign unter kinetischen Randbedingungen und legt damit den Grundstein für nachfolgende komplexere funktionelle Designs.
Kombination aus intelligenter Agentenproteingenerierung und inverser Normalschwingungsmodenkonstruktion
Die Forschung zur Generierung intelligenter Agentenproteine und zum inversen Design auf Basis normaler Schwingungsmodenformen entwickelt sich zu einem hochaktuellen Thema im Bereich des Protein-Engineerings und treibt sowohl die akademische Forschung als auch die industrielle Innovation voran.
In der akademischen Forschung haben zahlreiche Universitätsteams diesen Bereich kontinuierlich untersucht und eine Reihe bahnbrechender Ergebnisse erzielt. Einige Teams haben das kollaborative Framework intelligenter Agenten optimiert,Die Kombination der Analyse normaler Schwingungsmoden mit einem fortschrittlicheren Protein-Sprachdiffusionsmodell mildert das Entartungsproblem beim inversen Design effektiv.Diese Arbeit bestätigte zudem den intrinsischen Zusammenhang zwischen der Form der normalen Schwingungsmoden und der Sekundärstruktur sowie den dynamischen Eigenschaften von Proteinen und lieferte damit eine solidere theoretische Grundlage und einen besseren technischen Weg für die Neuentwicklung von Proteinen mit spezifischen Funktionen.
Ein anderes Team konzentrierte sich auf die Reduzierung des Speicherbedarfs und die Generalisierung, optimierte die Parametergröße und die Trainingsstrategie vortrainierter Protein-Sprachmodelle und entwickelte kleinere Modelle, die leichter zu generalisieren sind.Darüber hinaus wurde die Anwendung des inversen Designs von Normalschwingungsmoden auf spezifische Bereiche wie das Design katalytischer Zentren von Enzymen und die Optimierung von Proteinbindungsmitteln ausgedehnt.Dies hat eine solide Grundlage für die nachfolgende industrielle Transformation geschaffen.
Darüber hinaus hat Google DeepMind AlphaProteo auf den Markt gebracht.Als erstes Werkzeug künstlicher Intelligenz zur Entwicklung neuartiger, hochfester Proteinklebstoffe kann es neue Proteinkonjugate für eine Vielzahl von Zielproteinen generieren.Der Test, der auch den vaskulären endothelialen Wachstumsfaktor A (VEGF-A) umfasst, der mit Komplikationen von Krebs und Diabetes in Verbindung gebracht wird, erzielte eine höhere Erfolgsrate. Seine Bindungsaffinität ist 3- bis 300-mal höher als die der besten bestehenden Methoden. Dies dürfte die Entwicklung von Krebs- und Virostatika beschleunigen und liefert zudem neue Ansätze für die Entwicklung von Biosensoren und die Verbesserung der Insektenresistenz von Nutzpflanzen.
Andere Unternehmen konzentrieren sich auf die Schwachstellen der Arzneimittelentwicklung und nutzen die inverse Designtechnologie der normalen Schwingungsmodenform, um Proteinmedikamente für spezifische Krankheitsziele zu entwickeln. Dadurch wird der Entwicklungszyklus verkürzt, die Kosten werden gesenkt und die Entwicklung von Proteinmedikamenten in eine präzisere und effizientere Richtung gefördert.
Die kontinuierliche Optimierung der Designgenauigkeit und der Generalisierungsfähigkeit von Modellen durch die akademische Gemeinschaft sowie die laufenden Bemühungen der Industrie um eine effizientere Implementierung und breitere Anwendungsszenarien treiben die Proteindesign-Technologie gemeinsam in Richtung höherer Präzision, Effizienz und Vielfalt voran. Mit zunehmender Reife der Technologie werden Proteindesignmethoden, die auf intelligenten Agenten und der Analyse normaler Schwingungsmoden basieren, voraussichtlich in Bereichen wie der Pharmazie, der industriellen Produktion und der Bioproduktion breitere Anwendung finden und neue Durchbrüche ermöglichen.








