Command Palette
Search for a command to run...
Die Cornell University Schlägt Ein Innovatives KI-Framework Zur Entschlüsselung Des Chemischen Mechanismus Hochleitfähiger Lithium-Ionen-Elektrolyte Vor Und Erzielt Eine Vorhersagegenauigkeit Von Über 80 % Für %.

Angesichts der rasanten Expansion des Marktes für neue Energiebatterien, insbesondere der weitverbreiteten Anwendung von Lithium-Ionen-Batterien, Festkörperbatterien und Hochenergiebatterien, ist die Optimierung der Elektrolytleistung zu einem Schlüsselfaktor für die Sicherheit, Effizienz und Lebensdauer der Batterie geworden.
Die Salz-Lösungsmittel-Chemie ist die Grundlage für das Elektrolytverhalten in den meisten Lithium-Ionen-Batteriesystemen und bestimmt Schlüsseleigenschaften wie Ionenleitfähigkeit, Viskosität und chemische Stabilität. Ihre gezielte Entwicklung wird jedoch durch einen riesigen chemischen Raum mit unzähligen Kombinationen und nichtlinearen Struktur-Eigenschafts-Beziehungen erschwert. Spärliche und ungleichmäßig verteilte experimentelle Daten verschärfen dieses Problem zusätzlich und beeinträchtigen die Generalisierbarkeit von Modellen. In den letzten Jahren wurden zwar Fortschritte bei der KI-gestützten, autonomen Elektrolytentwicklung erzielt,Allerdings fehlt es der bestehenden Forschung noch immer eindeutig an einem einheitlichen Rechenparadigma, das die inhärente Interpretierbarkeit bewahren und gleichzeitig den riesigen chemischen Raum untersuchen kann, der von großtechnischen Elektrolytformulierungen abgedeckt wird.
In diesem ZusammenhangEin Forschungsteam der Cornell University hat mit SCAN ein robustes, interpretierbares und dateneffizientes Framework zur Modellierung und Interpretation der Salz-Lösungsmittel-Chemie entwickelt.Dieses Framework verarbeitet effektiv Daten mit langen Ausläufern und erfasst das gesamte Spektrum von Salz-Lösungsmittel-Formulierungen. Forscher wandten SCAN auf nicht-wässrige Elektrolytsysteme (NAE) an und erreichten einen Basisfehler von 0,372 mS·cm⁻¹ bei der Leitfähigkeitsvorhersage. Dies entspricht einer Reduzierung des Vorhersagefehlers um 65,31 TP³T im Vergleich zum Basismodell.
Noch wichtiger ist jedoch, dass groß angelegte Validierungsstudien zeigen, dassDas Modell erreichte eine Vorhersagegenauigkeit von 81,08% für die bestplatzierten Kandidatensysteme.Zusätzlich zu seinen Vorhersagefähigkeiten deckt SCAN den chemischen Mechanismus auf, indem es Gradientenentkopplung, symbolische Regression und quantenchemische Berechnungen einführt, um den Einfluss der molekularen Flexibilität und der Ion-Lösungsmittel-Wechselwirkungen auf die Leitfähigkeit zu ermitteln.
Die zugehörigen Forschungsergebnisse mit dem Titel „Ein dynamisches, routingbasiertes, interpretierbares Rahmenwerk für die Salz-Lösungsmittel-Chemie“ wurden in Nature Computational Science veröffentlicht.
Forschungshighlights:
* SCAN schließt eine wichtige Lücke in der Forschung zur Hochleistungs-NAE-Salz-Lösungsmittel-Chemie.
Inspiriert vom atomzentrierten Potentialenergieflächenmodell haben wir ein Multi-Feature-Netzwerk (MFNet) mit einer deskriptorzentrierten dedizierten Repräsentation und einem Aufmerksamkeitsmechanismus entwickelt.
* Innovativ wird in MFNet eine dynamische Routing-Strategie eingeführt, die es dem Modell ermöglicht, die Ionenleitfähigkeit über einen weiten Bereich genau vorherzusagen, ohne die ursprüngliche Datenverteilung zu verändern.

Papieradresse:
https://www.nature.com/articles/s43588-026-00955-5
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „SCAN“, um das vollständige PDF zu erhalten.
Die Datendimensionen werden umfassend abgedeckt.
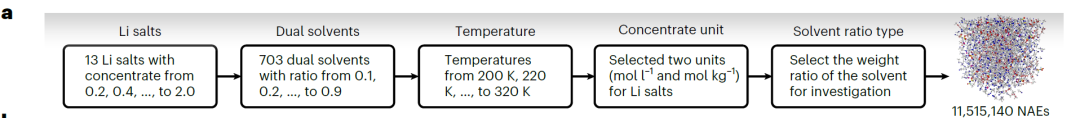
Um ein hochpräzises SCAN-Modell zu trainieren,Das Forschungsteam erstellte den CALiSol-Datensatz, der 13 Lithiumsalze und 38 organische Lösungsmittel umfasst (siehe Abbildung unten) und insgesamt 13.302 vollständige Datensätze enthält.
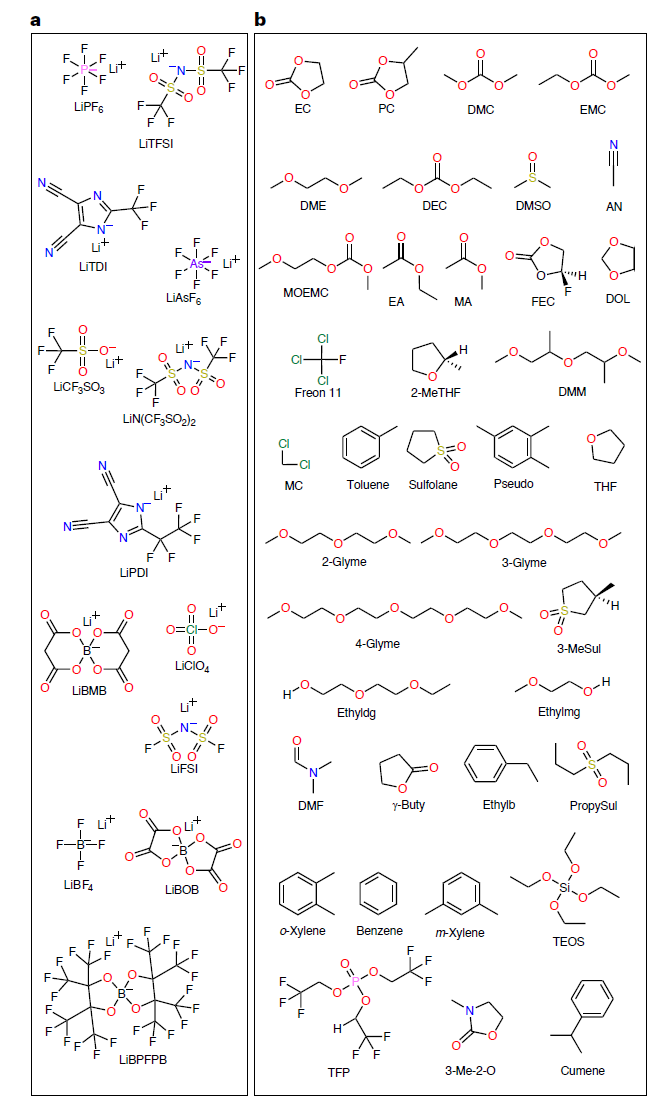
Die Datendimensionen sind umfangreich, und jeder Datenpunkt enthält Folgendes:
* Ionenleitfähigkeit k: 0–38,1 mS/cm
* Temperatur T: 194,15–477,42 K
* Salzkonzentration c: 0–4 mol/L oder mol/kg
* Lithiumsalztypen: LiPF₆, LiTFSI, LiFSI, LiBOB usw., insgesamt 13 Typen.
* Lösungsmittelarten: darunter Ethylencarbonat (EC), Methylethylcarbonat (EMC), Propionitril (AN) und viele andere.
* Gemischte Strategie SRT: Mol-, Volumen- oder Massenverhältnis
Die Molekülinformationen aller Lithiumsalze und Lösungsmittel wurden mithilfe von SMILES-Sequenzen in dreidimensionale Molekülkoordinaten umgewandelt und eine Geometrieoptimierung auf dem theoretischen B3LYP/6-31G-Niveau durchgeführt, um eine genaue und zuverlässige Molekülstruktur und elektronische Eigenschaften zu gewährleisten. Durch diese Methode wurdeDer Datensatz liefert nicht nur Werte für die Ionenleitfähigkeit, sondern auch molekulare Eigenschaften, Strukturinformationen und die Solvatationsumgebung für jedes System.Liefern Sie aussagekräftige Eingangsdaten für KI-Modelle und achten Sie dabei auf ein ausgewogenes Verhältnis zwischen Datenintegrität und wissenschaftlicher Interpretierbarkeit.
Bei der Erstellung des Datensatzes legte das Forschungsteam besonderes Augenmerk auf das Problem der Long-Tail-Daten (LTD): Die Anzahl der hochleitfähigen Systeme ist begrenzt, während die Leitfähigkeit der meisten Systeme niedrig ist - k < 5 mS·cm⁻¹ für 9.115 NAEs (etwa 68,51 TP3T) und k > 20 mS·cm⁻¹ nur für 67 (etwa 0,51 TP3T).
SCAN: Nutzt MFNet und dynamische Routing-Mechanismen
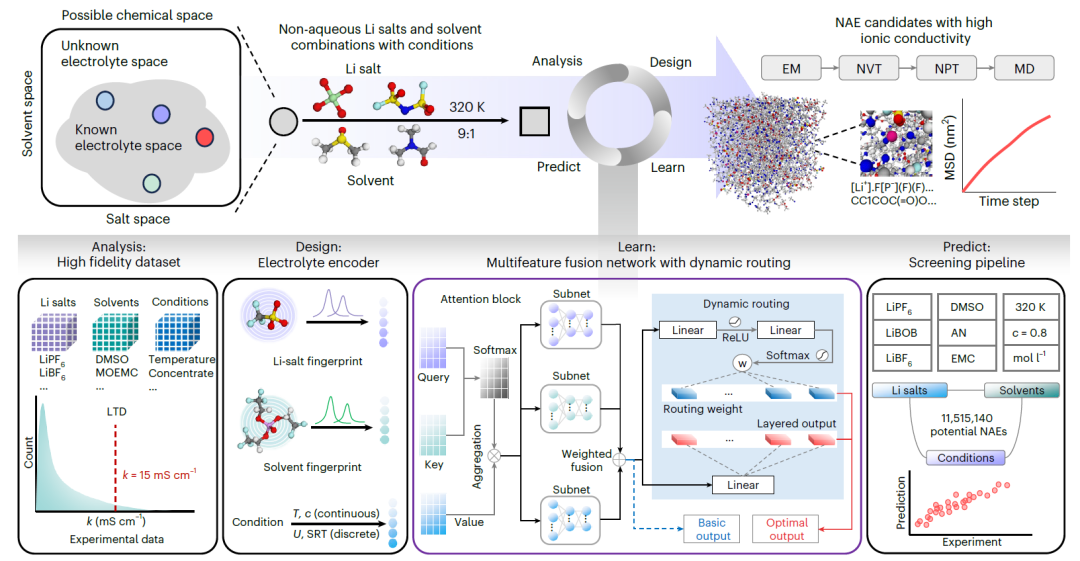
SCAN ist eine NAE-Engineering-Plattform mit dynamischem Routing. Das folgende Diagramm veranschaulicht den gesamten Workflow:
Analyse
Es wurde ein hochpräziser experimenteller Datensatz erstellt, der eine Vielzahl von Kombinationen aus Lithiumsalzen, Lösungsmitteln und Versuchsbedingungen abdeckt. Jede Variable wurde statistisch analysiert, einschließlich der Charakterisierung der langschwänzigen Verteilung der k-Werte.
Design
Für Lithiumsalze bzw. Lösungsmittel wurden molekulare Fingerabdrücke mit chemischen Informationen entwickelt und mit bedingter Codierung kombiniert, um Salz-Lösungsmittel-Bedingungs-Kombinationen genau zu charakterisieren.
Lernen
Zur Vorhersage des k-Werts von NAEs wurde ein maßgeschneidertes Multi-Feature-Fusionsnetzwerk (MFNet) entwickelt. Unabhängige Aufmerksamkeitsmodule wurden erstellt, indem Anfrage-, Schlüssel- und Wertdaten eingebettet wurden, um die Repräsentationen von Salz, Lösungsmittel und Bedingung zu verarbeiten. Anschließend wurden diese mithilfe eines vollständig verbundenen neuronalen Netzwerks fusioniert. Zusätzlich wurde eine dynamische Routing-Strategie eingeführt, um die Modellleistung und -robustheit zu verbessern.
Vorhersagen
Zur Identifizierung potenzieller NAEs aus über zehn Millionen Salz-Lösungsmittel-Kombinationen wurde ein Hochdurchsatz-Screening-Verfahren entwickelt. Kandidatensysteme mit hohen vorhergesagten k-Werten wurden anschließend mittels Molekulardynamik-Simulationen (MD) validiert. Diese umfassten Energieminimierung (EM), Simulationen im kanonischen Ensemble (NVT), im isothermen-isobaren Ensemble (NPT), MD-Produktionssimulationen, Analysen der mittleren quadratischen Verschiebung (MSD) und Berechnungen der radialen Verteilungsfunktion (RDF).
MFNet und der dynamische Routing-Mechanismus bilden den Kern des gesamten Frameworks, und die spezifischen Mechanismen sind wie folgt:
MFNet: Multichannel Self-Attention Network
Inspiriert vom atomaren Zentralpotentialflächenmodell unterteilt das MFNet-Framework (Molecular Feature Network) das Netzwerk in drei unabhängige Teilnetze, die jeweils unterschiedliche Funktionen übernehmen:
* Lithium-Salz-Subnetz: Im CALiSol-Datensatz enthält jedes NAE nur einen Typ von Lithiumsalz, daher wird sein Deskriptorvektor direkt in das Lithium-Salz-Subnetz eingegeben.
* Lösungsmittel-Subnetz: Da einige Datenpunkte mehrere Lösungsmittel umfassen (z. B. PC und AN im Verhältnis 0,9:0,1 gemischt), wird der Mittelwert dieser Lösungsmitteldeskriptoren als Eingabe berechnet, um die gesamte Lösungsmittelumgebung widerzuspiegeln.
* Bedingtes Subnetz: Verarbeitet experimentelle Bedingungen wie Temperatur und Konzentration.
Im Anschluss an das Selbstaufmerksamkeitsmodul werden zwei vollständig verbundene verborgene Schichten für die progressive Projektion und nichtlineare Transformation der Eingabe verwendet. Dadurch werden hochdimensionale Merkmale generiert, um die Ausdrucksstärke der nachfolgenden Verarbeitung zu verbessern. Zwischen den Schichten wird die nichtlineare ReLU-Aktivierungsfunktion eingesetzt, um die Repräsentationsfähigkeit des Modells weiter zu steigern.
Die finale Netzwerkarchitektur sieht wie folgt aus: ein Lithiumsalz-Subnetz (14–16–16), ein Lösungsmittel-Subnetz (14–16–16) und ein bedingtes Subnetz (6–16–16). Potenzielle Ausgaben mit intrinsischen Dimensionen (Merkmalsdimensionen < 128) werden einem gewichteten Fusionsmodul zugeführt, um gewichtete Ausgaben zu berechnen und dabei die globalen Abhängigkeitsinformationen der Eingabemerkmale zu erhalten. Da die Kodierung von Lithiumsalz-, Lösungsmittel- und bedingten Merkmalen unabhängig von drei Subnetzen verarbeitet wird, kann Single-Head Self-Attention (SHA) zur ressourcenschonenden Verarbeitung kleiner Merkmalsdaten (jeweils 14, 14 und 6 Dimensionen) eingesetzt werden. Dies macht SHA besonders geeignet für Aufgaben mit niedrigen Einbettungsdimensionen.
Dynamischer Routing-Mechanismus: Lösung des Problems von Long-Tail-Daten
In einer Verteilung mit langem Ausläufer konzentrieren sich die Datenpunkte hauptsächlich im „Kopfbereich“, während Datenpunkte im „Ausläuferbereich“ selten sind. Traditionelle Modelle neigen jedoch dazu, die Datenpunkte im Kopfbereich zu überanpassen und den Ausläuferbereich zu vernachlässigen, was zu einer unzureichenden Erforschung des kritischen, aber knappen chemischen Raums führt. Um dieses Problem zu lösen, führt diese Studie eine dynamische Routing-Strategie in MFNet ein. Im Gegensatz zur Standardarchitektur, die alle Datenpunkte gleich behandelt,Das dynamische Routing lernt einen Soft-Gating-Mechanismus, um verschiedenen Schichten adaptiv unterschiedliche Repräsentationsfähigkeiten basierend auf der Schichteingabe zuzuweisen.Wie unten gezeigt:

Dieser Mechanismus ermöglicht es seltenen Stichproben, unterschiedliche Routing-Pfade zu aktivieren und bedingte Berechnungen zu erleichtern, wodurch die Generalisierungsfähigkeit für Kategorien mit niedriger Frequenz verbessert wird. Seine zwei Hauptmerkmale sind:
* Eingangsabhängige Routing-Gewichte: das heißt, Auswahl eines Merkmalsunterraums für jede Stichprobe, ohne die ursprüngliche Verlustfunktion oder Datenverteilung zu verändern;
* Kategorieadaptive Feature-Entkopplung: Hierbei wird der Unterschied zwischen der dominanten Kategorie und den Randkategorien explizit modelliert. Dies zeigt, dass dynamisches Routing eine flexiblere und besser interpretierbare LTD-Lösung bietet als einfaches statisches Loss Reshaping.
Interpretierbarkeit: GBA und symbolische Regression
SCAN ermöglicht die chemische Interpretierbarkeit und sagt gleichzeitig die Ionenleitfähigkeit präzise voraus:
* GBA (Gradientenbasierte Attribution):Da das SCAN-Modell ein neuronales Netzwerk mit drei parallelen Aufmerksamkeitsmechanismen verwendet, ist sein Entscheidungsprozess im Vergleich zu Baummodellen komplexer zu visualisieren. Um die wichtigsten chemischen Faktoren zu identifizieren, die k beeinflussen, wurde die GBA-Methode zur Bewertung der Merkmalswichtigkeit eingesetzt. Dabei wurde der Gradientenbeitrag jedes Eingangsmerkmals zum Modelloutput berechnet und die wichtigsten Lithiumsalz-, Lösungsmittel- und bedingten Merkmale identifiziert.
* Symbolische Regression:Um einen interpretierbaren funktionalen Zusammenhang zwischen wichtigen Li-Salz-, Lösungsmittel- und Bedingungsinformationen und kkk zu ermitteln, wurde eine symbolische Regressionsmethode auf Basis von PySRRegressor verwendet.
Das SCAN-Modell übertrifft alle Vergleichsmodelle durchweg.
Um die Leistungsfähigkeit des SCAN-Frameworks unter Verwendung von MFNet und dynamischem Routing umfassend zu bewerten, haben die Forscher vier auf MFNet basierende Basismodelle konstruiert.Keines davon beinhaltet dynamisches Routing: (1) Grundlegendes MFNet-Modell: B1 (unverarbeiteter Deskriptor) und B2(Deskriptor skaliert nach Maximalwert); (2) MFNet-Modell mit Oversampling-Technik für Minderheitsklassen SMOTE: S1-S18(3) MFNet-Modell in Kombination mit der K-Nearest-Neighbor-Undersampling-Technik (KUTE): U1–U9(4) MFNet-Modell, das SMOTE und KUTE kombiniert: SU1–SU9 .
Gleichzeitig wurden Transformer-basierte Modelle, Graph Neural Networks (GNNs), Support Vector Regression (SVR) und Extreme Gradient Boosting (XGBoost) als Vergleichsgrundlage herangezogen (siehe Abbildung unten). Darüber hinaus wurden SCAN-Modelle mit Single-Head Attention (SHA) oder Multi-Head Attention (MHA) und unterschiedlicher Anzahl dynamischer Routing-Schichten (η = 2–10) implementiert, um die Robustheit der Modelle zu überprüfen.
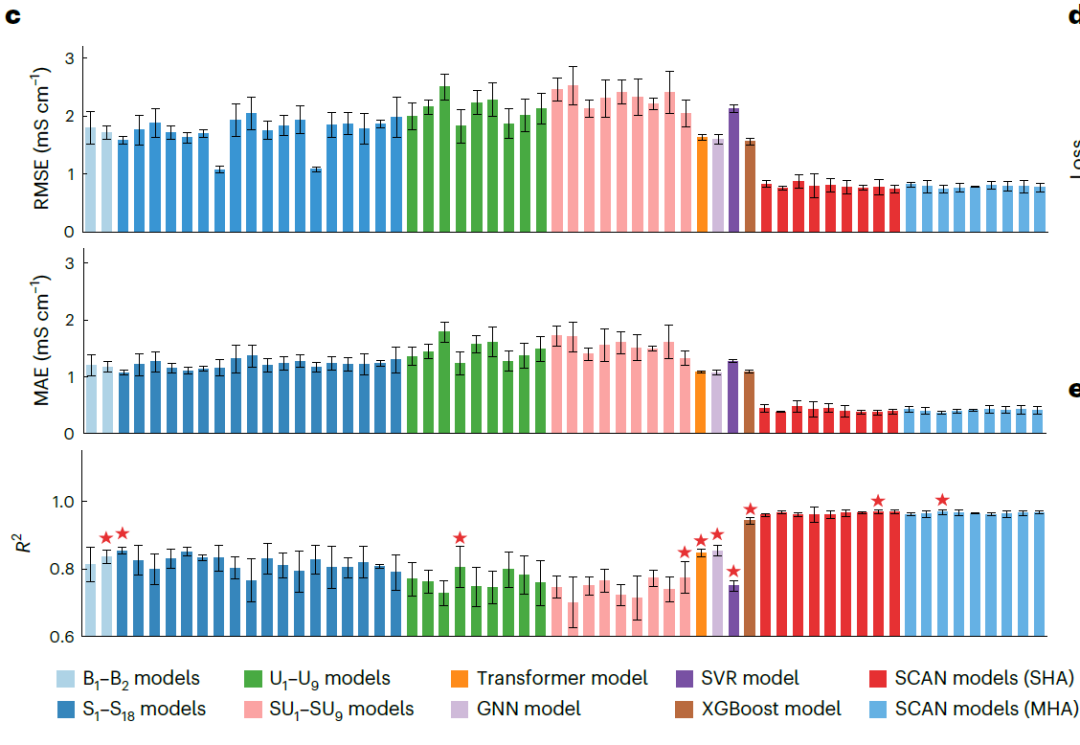
NAE-Leistungsbewertung
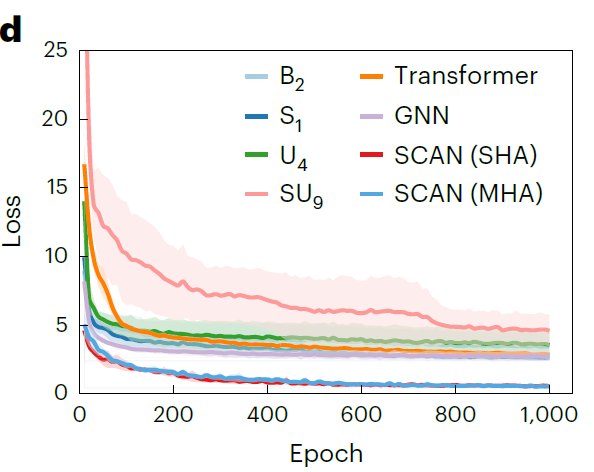
Die in der folgenden Abbildung dargestellten Ergebnisse zeigen, dass das SCAN-Modell mit SHA (η = 9) alle Basismodelle durchweg übertrifft und deutlich geringere Vorhersagefehler (RMSE 0,769 mS·cm⁻¹, MAE 0,372 mS·cm⁻¹) sowie ein höheres R² (0,969) aufweist. Es ist anzumerken, dass…Im Vergleich zum leistungsstärksten Baseline-GNN (1,072 mS·cm⁻¹) in MAE wurde der Fehler um 65,31 TP3T reduziert.Die SCAN-Performance unter Verwendung von MHA (η = 4) ist mit der der SHA-Version vergleichbar, was darauf hindeutet, dass MFNet eine gute Robustheit bei der Integration mit dynamischem Routing aufweist.
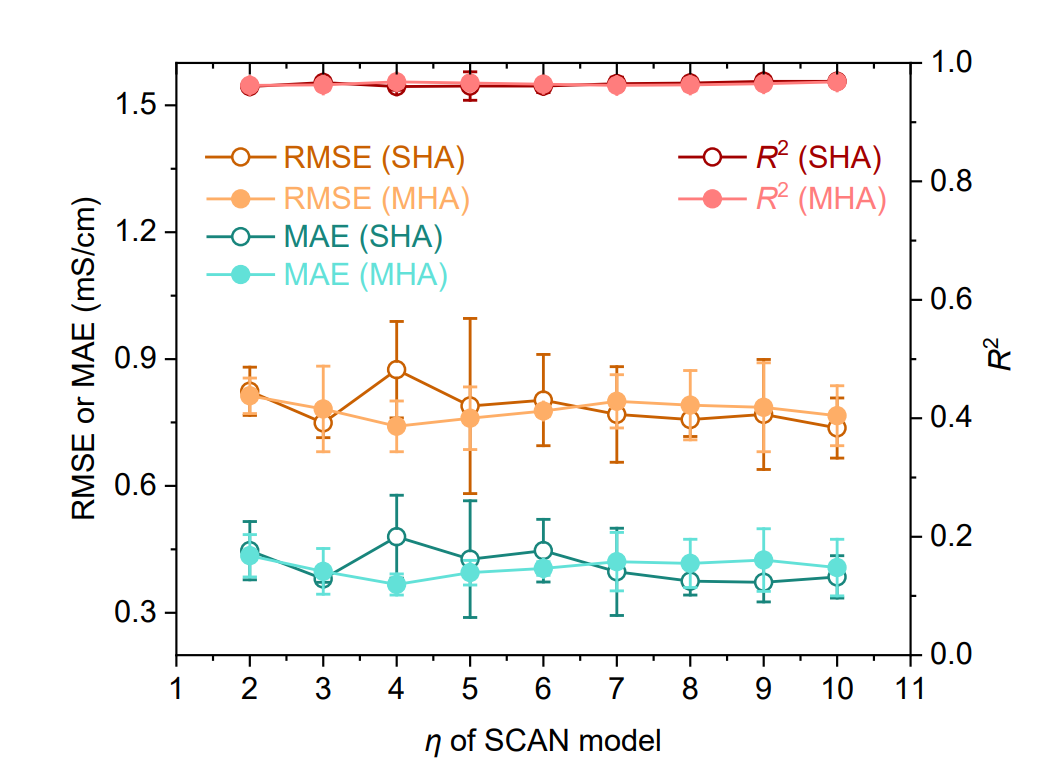
Auch,Die Forscher stellten die Validierungsverlustkurven von SCAN und eines repräsentativen Basismodells über 1.000 Trainingsepochen grafisch dar.Wie aus der untenstehenden Abbildung hervorgeht, ist der Vorhersagefehler von SCAN von Beginn des Trainings an deutlich geringer als der des Basismodells, mit einem anfänglichen Verlust von nur 4,59 mS·cm⁻¹, und sein Validierungsverlust sinkt weiter und konvergiert auf ein noch niedrigeres Niveau, was darauf hindeutet, dass es eine stärkere Stabilität und Generalisierungsfähigkeit besitzt.
Hochdurchsatz-NAE-Screening-Fähigkeit
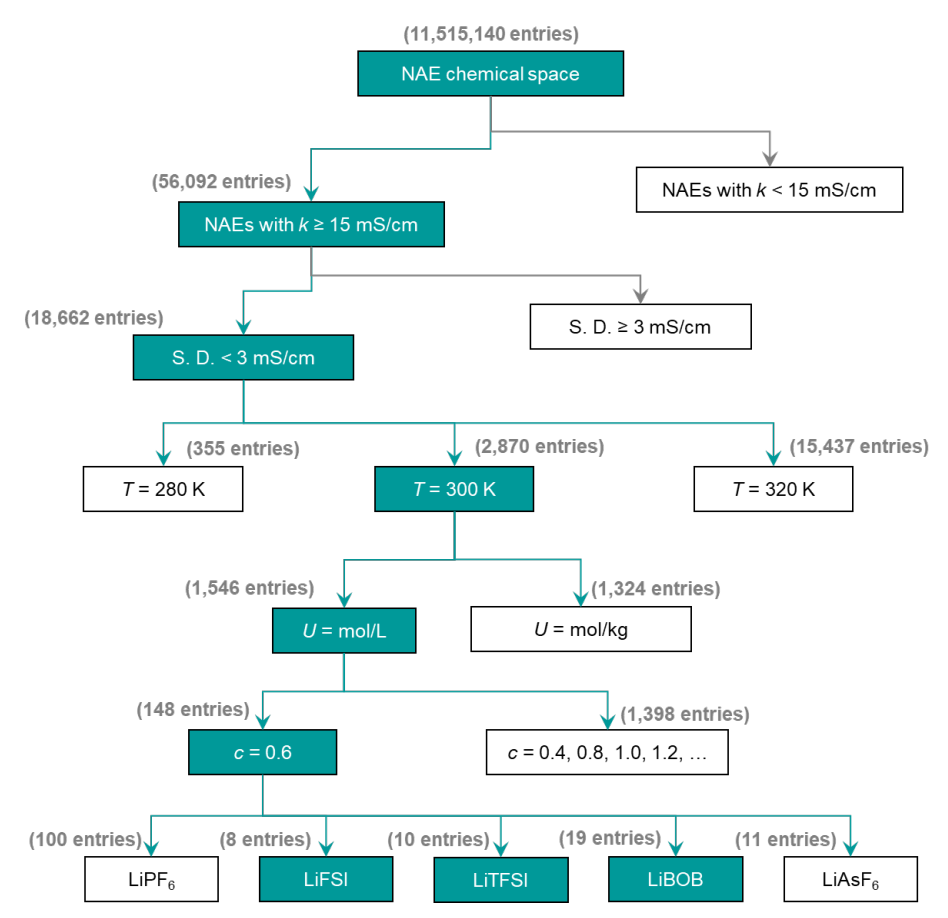
Hochpräzise Scans ermöglichen die effiziente Erforschung des erweiterten chemischen Raums von NAEs. Zu diesem Zweck haben Forscher ein Hochdurchsatz-Screeningverfahren entwickelt (siehe Abbildung unten), das darauf abzielt, NAEs mit hoher k-Wert zu identifizieren.Auf dieser Grundlage wurden 11.515.140 potenzielle Dual-Solvent-NAEs generiert.
Durch die Nutzung der Vorhersagekraft von SCAN konnten 56.092 NAEs mit k ≥ 15 mS·cm⁻¹ schnell identifiziert werden, von denen 18.662 eine geringe Vorhersageunsicherheit (< 3 mS·cm⁻¹) aufwiesen. Dieses Verfahren reduziert den Rechenaufwand und die Kosten von Molekulardynamiksimulationen und -experimenten erheblich.
Validierung von Kandidaten für nicht-wässrige Elektrolyte (NAEs)
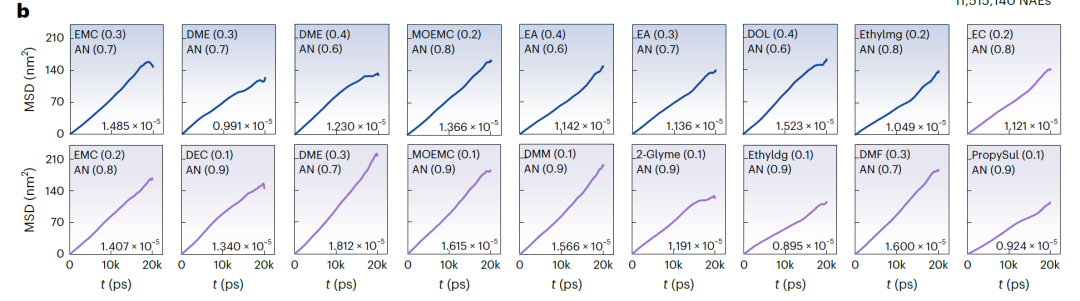
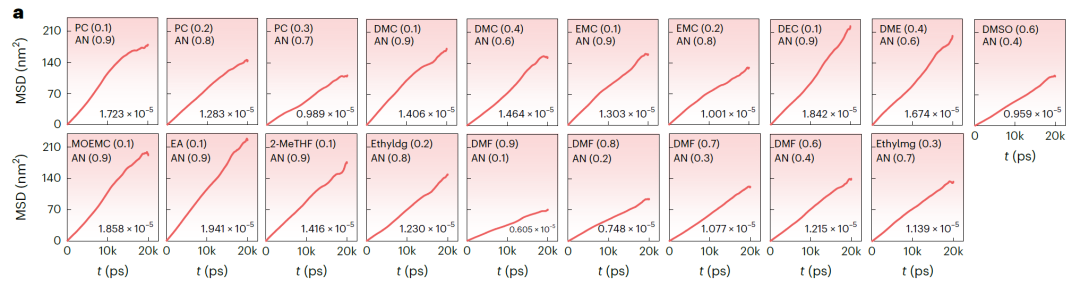
Um die Vorhersagen rigoros zu validieren, führten die Forscher Molekulardynamik-Simulationen (MD) durch, um den k-Wert zu erhalten. Basierend auf Temperatur (T), Konzentration (c) und den tatsächlichen Bedingungen beschränkten sie den Umfang der MD-Validierung auf NAE-Kandidatensysteme auf Basis von LiFSI, LiTFSI und LiBOB und wählten schließlich 37 vielversprechende Systeme für eine detaillierte Untersuchung aus, wie in der folgenden Abbildung dargestellt:
Die durchschnittliche Rechenzeit für jedes System beträgt etwa 10 Stunden (36.355,12 Sekunden), und die geschätzten Kosten für die Simulation von 10⁷ Kandidatensystemen belaufen sich auf etwa 10⁸ GPU-Stunden – was die praktischen Möglichkeiten einer rein methodischen Auswahl weit übersteigt, selbst unter idealen Hochleistungsrechnerbedingungen. Im Vergleich dazuEin gut trainiertes SCAN-Modell kann den gesamten Prozess von der Deskriptorberechnung bis zur endgültigen Vorhersage für jedes Kandidatensystem in weniger als 5 Sekunden abschließen und so die Rechenkosten um mehr als das 7.200-fache reduzieren.Dies verbessert Skalierbarkeit und Effizienz erheblich. Daraus wird die Notwendigkeit des SCAN-Frameworks bei der NAE-Entdeckung unterstrichen, dessen Ersatzmodell leistungsstarke Kandidaten schnell und mit extrem geringem Rechenaufwand priorisieren kann.
Abbildung b (NAEs basierend auf LiFSI und LiTFSI) und Abbildung a (NAEs basierend auf LiBOB) zeigen die mittlere quadratische Verschiebung (MSD) über die Zeit und den entsprechenden Diffusionskoeffizienten aus MD-Simulationen. Die MSD aller Systeme zeigt einen stabilen linearen Anstieg, was auf eine stabile und konvergente Simulation hindeutet. Die mit SCAN vorhergesagten k-Werte stimmen sehr gut mit den MD-Simulationsergebnissen überein, mit einer durchschnittlichen Abweichung von nur 3,198 mS·cm⁻¹ und einer maximalen Abweichung von 7,342 mS·cm⁻¹. In den Validierungssystemen betrug die Erfolgsrate für 25 Systeme mit k > 15 mS·cm⁻¹ 67,571 TP⁻¹. Für Systeme mit vorhergesagtem k > 14 mS·cm⁻¹ stieg die Validierungserfolgsrate auf 81,081 TP⁻¹.
Künstliche Intelligenz verändert das grundlegende Paradigma der Batterieforschung und -entwicklung.
Batterien sind als Kernkomponente in wichtigen Anwendungen wie Unterhaltungselektronik, Elektrofahrzeugen und Energiespeichersystemen die treibende Kraft der globalen Energiewende. Die Verbesserung der Energie- und Leistungsdichte, die Verlängerung der Lebensdauer, die Erhöhung der Sicherheit und die Senkung der Herstellungskosten sind die zentralen Ziele der Batterieforschung und -entwicklung. Der Schlüssel zum Erreichen dieser Ziele liegt in einem tiefen Verständnis der elektrochemischen Mechanismen in Batterien, insbesondere der elektrochemischen Grenzflächenreaktionen und Stabilisierungsmechanismen, der Kopplung und des Transports von Elektronen und Ionen sowie der Energiespeichermechanismen von Elektrodenmaterialien der nächsten Generation.
Aus einer breiteren Perspektive der technologischen Entwicklung wird das durch das SCAN-Framework repräsentierte Paradigma der „datengetriebenen und interpretierbaren Modellierung“ zu einem wichtigen Bestandteil des Batterie-Forschungs- und Entwicklungssystems der nächsten Generation. In den letzten Jahrzehnten basierte die Innovation bei Batteriematerialsystemen hauptsächlich auf Erfahrungswerten und Versuch-und-Irrtum-Experimenten, was zu langen Entwicklungszyklen und hohen Kosten führte. Durch die Integration von maschinellem Lernen und Hochleistungsrechnen wird die Batterie-Forschung und -Entwicklung jedoch zunehmend effizienter.
Zum Beispiel,Wen Yan und Sheng Gong sowie weitere Mitglieder des Seed-Teams von ByteDance entwickelten ein einheitliches Rahmenwerk für die Formulierung von Elektrolyten, das Vorwärtsvorhersagemodelle mit Rückwärtsgenerierungsmethoden integriert.Forscher trugen umfangreiche Literaturdaten zu einzelnen Molekülen (über 240.000) und Molekülmischungen (über 10.000) mit Eigenschaftskennzeichnungen zusammen und deckten damit den Designraum von Elektrolyten umfassend ab. Durch die Einbeziehung von über 100.000 Daten zu Molekülmischungen aus Molekulardynamiksimulationen konnten sie ein präzises Modell für maschinelles Lernen trainieren, das nicht nur die Leitfähigkeit, sondern auch Schmelzstrukturen im Zusammenhang mit der Grenzflächenstabilität von Lithium-Metall-Batterien vorhersagen kann.
Titel des Beitrags: Eine einheitliche prädiktive und generative Lösung für die Formulierung flüssiger Elektrolyte
Link zum Artikel:
https://www.nature.com/articles/s42256-025-01173-w
Insgesamt verändert künstliche Intelligenz das grundlegende Paradigma der Batterieforschung und -entwicklung. Durch die tiefgreifende Integration von Modellen und experimentellen Systemen wird erwartet, dass die Geschwindigkeit der Entdeckung und die Innovationsdichte von Batteriematerialien einen neuen, beschleunigten Zyklus erreichen werden.
Quellen:
https://www.nature.com/articles/s43588-026-00955-5
https://phys.org/news/2026-02-ai-framework-reveals-chemistry-high.html
https://static-content.springer.com/esm/art%3A10.1038%2Fs43588-026-00955-5/MediaObjects/43588_2026_955_MOESM1_ESM.pdf
https://www.eet-china.com/mp/a471613.html








