Command Palette
Search for a command to run...
Die Columbia University Und Die Stanford University Kooperieren! Squidiff Ermöglicht Die Simulation Des Transkriptoms in Verschiedenen Szenarien Und Trägt so Zur Entwicklung Der Präzisionsmedizin Und Der Weltraummedizin bei.
In der Zellbiologie sind lebende Zellen stets komplexe, dissipative Systeme, die sich weit vom chemischen Gleichgewicht entfernt befinden. Wie ihre kollektive Reaktion auf äußere Reize ausfällt, ist nach wie vor eine zentrale wissenschaftliche Frage, die Forscher zu beantworten suchen. Diese Reaktion wird nicht nur durch die Heterogenität des Gewebes und externe Signale gemeinsam reguliert, sondern weist häufig auch unvorhersehbare, nichtlineare dynamische Eigenschaften auf. Obwohl die Einzelzellsequenzierung eine unvoreingenommene Analyse der heterogenen Zellzusammensetzung ermöglicht, stellt die präzise Erfassung der Veränderungen im gesamten Transkriptom nach einer Stimulation weiterhin eine große Herausforderung dar.
Um diese Einschränkung zu überwinden, hat die wissenschaftliche Gemeinschaft bereits verschiedene Modelle des maschinellen Lernens wie scGen und CellOT entwickelt. Diese Modelle schneiden jedoch bei der Vorhersage hochauflösender dynamischer Übergänge schlecht ab und basieren größtenteils auf aufgabenspezifischen Designs, was ihre Anwendbarkeit stark einschränkt. Das Aufkommen von Diffusionsmodellen hat in diesem Bereich einen neuen Durchbruch gebracht: Durch die iterative Generierung optimierter Daten lassen sich reichhaltigere Datenverteilungseigenschaften erfassen, wodurch ein neuer Ansatz zur Lösung der genannten Probleme entsteht. Aktuell versuchen einige Studien, Diffusionsmodelle mit Variations-Autoencodern (VAEs) zu kombinieren oder den Diffusionsprozess im latenten Raum zu implementieren. Dadurch konnten erfolgreich hochpräzise Einzelzelldaten generiert und die Modellierungseffizienz verbessert werden.Die Anwendung von Diffusionsmodellen in Schlüsselszenarien wie der Vorhersage von Genperturbationsreaktionen, der Vorhersage von Arzneimittelperturbationsreaktionen und der Ableitung von Zellentwicklungstrajektorien ist jedoch nach wie vor ein unterentwickeltes Gebiet..
In diesem ZusammenhangForschungsteams der Columbia University, der Stanford University und anderer Universitäten haben das Squidiff-Rechenframework entwickelt.Dieses Framework basiert auf einem bedingt entrauschten impliziten Diffusionsmodell und kann transkriptomische Reaktionen verschiedener Zelltypen unter Differenzierungsinduktion, Genperturbation und Arzneimittelbehandlung vorhersagen.Sein Hauptvorteil liegt in seiner Fähigkeit, definitive Informationen aus Gen-Editierungswerkzeugen und Wirkstoffen zu integrieren:Bei der Vorhersage der Stammzelldifferenzierung kann Squidiff nicht nur transiente Zellzustände präzise erfassen, sondern auch nicht-additive Genperturbationseffekte und zellspezifische Reaktionscharakteristika identifizieren. Das Forschungsteam wandte Squidiff zudem in der Forschung an vaskulären Organoiden an und konnte so erfolgreich die Auswirkungen von Strahlenexposition auf verschiedene Zelltypen vorhersagen und die Schutzwirkung von Strahlenschutzmedikamenten bewerten.
Die zugehörigen Forschungsergebnisse mit dem Titel „Squidiff: Vorhersage der zellulären Entwicklung und Reaktionen auf Störungen mithilfe eines Diffusionsmodells“ wurden in Nature Methods veröffentlicht.

Papieradresse:
https://www.nature.com/articles/s41592-025-02877-y
Folgen Sie unserem offiziellen WeChat-Account und antworten Sie im Hintergrund mit „Squidiff“, um das vollständige PDF zu erhalten.
Weitere Artikel zu den Grenzen der KI:
https://hyper.ai/papers
Datensatz: Vollständige Abdeckung mehrerer Szenarien + standardisierte Qualitätskontrolle
Um die Leistungsfähigkeit des Squidiff-Frameworks vollständig zu trainieren und zu validieren,Das Forschungsteam erstellte einen Multi-Szenario-Datensatz, der sowohl simulierte als auch reale experimentelle Daten umfasst und wichtige Forschungsrichtungen wie Zelldifferenzierung, Genperturbation, medikamentöse Behandlung und Strahlenreaktion von vaskulären Organoiden abdeckt.Alle Daten durchliefen eine einheitliche Qualitätskontrolle: Zellen mit geringer Qualität (mitochondriales Genverhältnis > 20% oder < 1000 Gene) wurden herausgefiltert, Gene mit geringer Expression entfernt und in einigen Fällen zusätzlich Gene, die mit zwei Zelltypen oder Stress in Zusammenhang stehen, ausgeschlossen. Abschließend wurden Unterschiede in der Sequenziertiefe mittels Log-Normalisierung korrigiert, um die Vergleichbarkeit der Datensätze zu gewährleisten.
Bezüglich der simulierten Daten verwendete das Team das Splatter-Tool auf Basis einer hierarchischen Gamma-Poisson-Verteilung, um synthetische Einzelzell-RNA-Sequenzierungsdaten zu generieren, die die Expressionsheterogenität und Varianzcharakteristika realer scRNA-seq simulieren, um die grundlegenden Fähigkeiten des Modells bei der Transkriptomrekonstruktion und -inferenz ohne die Notwendigkeit einer zusätzlichen biologischen Vorverarbeitung zu überprüfen.
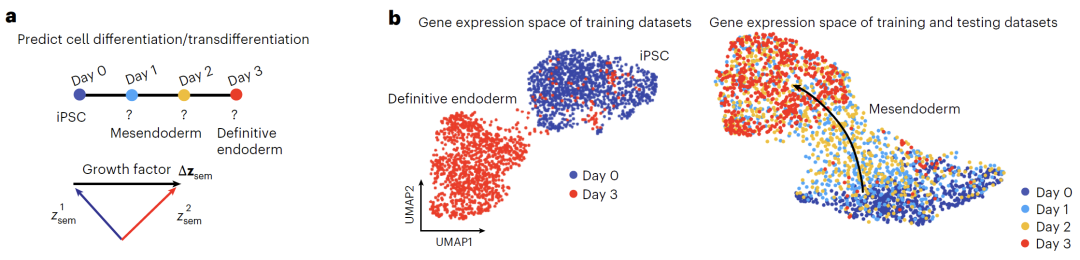
Die Daten zur Zelldifferenzierung stammen aus einem öffentlich zugänglichen Datensatz zur Differenzierung humaner induzierter pluripotenter Stammzellen (iPS-Zellen) in das Endoderm. Dieser Datensatz enthält die Transkriptome von 4.800 Zellen von Tag 0 (iPS-Zell-Zustand) bis Tag 3 (definierter Endoderm-Zustand). Das Modell nutzte die Daten von Tag 0 und Tag 3 als Trainingsdatensatz und die Daten von Tag 1 und Tag 2 als Testdatensatz. Die 203 hypervariablen Gene wurden für die Modellierung ausgewählt. Während des Trainings wurde Gaußsches Rauschen eingeführt und 1.000 Diffusionsschritte festgelegt. Die semantischen Variablen der Differenzierung wurden durch Berechnung der durchschnittlichen Differenz latenter Repräsentationen ermittelt. Anschließend wurde die Entwicklungstrajektorie von Tag 0 bis Tag 3 mittels linearer Interpolation simuliert, um die Vorhersagekraft des Modells für den dynamischen Differenzierungsprozess zu evaluieren.
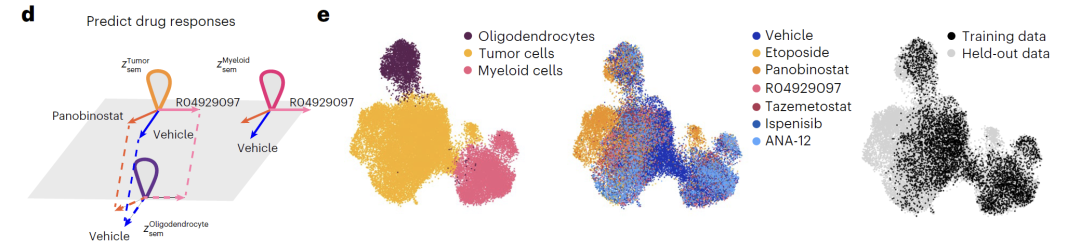
Die Daten zur Genperturbation stammen aus einem CRISPR-Screening-Experiment an K562-Zellen.Die Studie umfasste ca. 10.000 Zellen, darunter sowohl ZBTB25- als auch PTPN12-Gen-Knockout-Zellen sowie deren Wildtyp-Kontrollen. Die Daten wurden in drei Gruppen unterteilt: „PTPN12 + Kontrolle“, „ZBTB25 + Kontrolle“ und „PTPN12 + ZBTB25“. Die ersten beiden Gruppen dienten dem Training, die letzte Gruppe dem Testen. Nach dem Training wurden genspezifische Variablen extrahiert und kombiniert, um transkriptomische Veränderungen infolge kombinierter Doppelgenperturbation zu simulieren und so die Fähigkeit des Modells zur Erfassung nicht-additiver Effekte zu validieren.
Die Daten zur Arzneimittelverarbeitung integrieren mehrere Zell- und Arzneimittelproben.Dies umfasst Expressionsprofile von Glioblastomen, die mit sechs verschiedenen Medikamenten, darunter Etoposid, behandelt wurden, sowie Daten zum Ansprechen von Melanomen auf Medikamentenkombinationen. Während des Trainings lernt das Modell spezifische Perturbationsdarstellungen für jedes Medikament und integriert unbekannte Medikamentenproben aus dem sci-Plex3-Datensatz. Durch die Kombination von SMILES-Struktur, Dosierungsinformationen und Wirkstoff-Fingerprints ermöglicht es eine generalisierte Vorhersage der Perturbationseffekte unbekannter Medikamente.
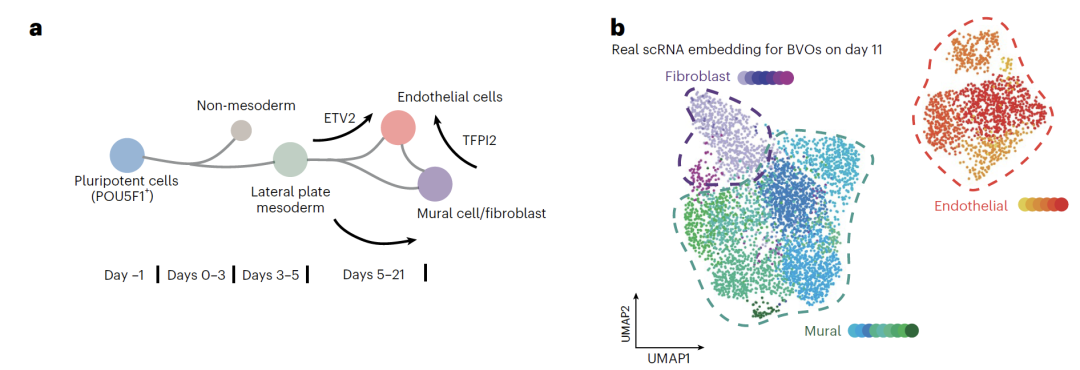
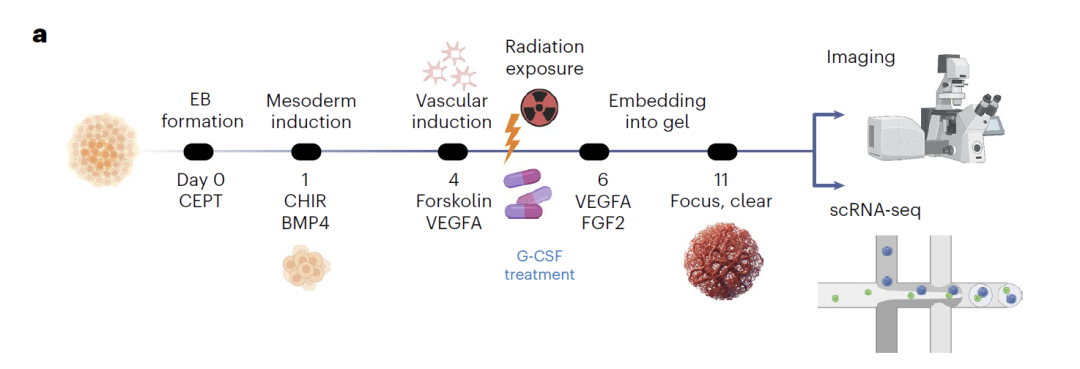
Die Daten zu den vaskulären Organoiden basieren auf eigenen Experimenten.Endothelzellen, Parietalzellen und Fibroblasten wurden aus gesunden humanen iPS-Zellen differenziert. Am fünften Tag wurden die Zellen Neutronen- oder Photonenstrahlung ausgesetzt, und am elften Tag wurden scRNA-Seq-Daten erhoben. So entstand eine Ressourcenbibliothek mit 72 Organoiden und ca. 60.000 Zellen. Die multimodale Validierung erfolgte zusätzlich durch ELISA-Messungen von Entzündungsfaktoren. Für die Modellierung nutzte das Team Daten vom ersten und elften Tag, um das Modell zu trainieren und durch Interpolation Zellzustände zu Zwischenzeitpunkten vorherzusagen. In den Szenarien mit Bestrahlung und G-CSF-Behandlung wurden ausschließlich Endothelzelldaten für das Training verwendet, wodurch veränderte Transkriptome für alle drei Zelltypen generiert wurden. Abschließend wurde die biologische Signifikanz der vorhergesagten Ergebnisse durch differentielle Expressions- und pseudotemporale Analysen validiert.
Squidiff: Ein bedingtes Diffusionsmodell, das DDIM und semantische Kodierung integriert
Um die dynamische Reaktion des Transkriptoms unter verschiedenen Störungen wie Differenzierung, Entwicklung, Genomeditierung und medikamentöser Behandlung präzise vorherzusagen, entwickelte das Forschungsteam Squidiff, ein intelligentes Rechenframework, das auf dem bedingten Diffusionsmodell basiert.Wie in der Abbildung unten dargestellt, integriert dieses Modell das Conditional Denoising Diffusion Implicit Model (DDIM) tiefgreifend mit semantischer Kodierungstechnologie, um eine dreistufige kollaborative Architektur aus „Kodierung-Diffusion-Dekodierung“ zu konstruieren. Es kann nicht nur effizient Transkriptomdaten generieren, die dem biologischen Hintergrund entsprechen, sondern auch den Zellzustand flexibel durch latente Variablen regulieren und ist breit anwendbar in verschiedenen Forschungsszenarien wie Zelldifferenzierung, Genperturbation und Arzneimitteltherapie.
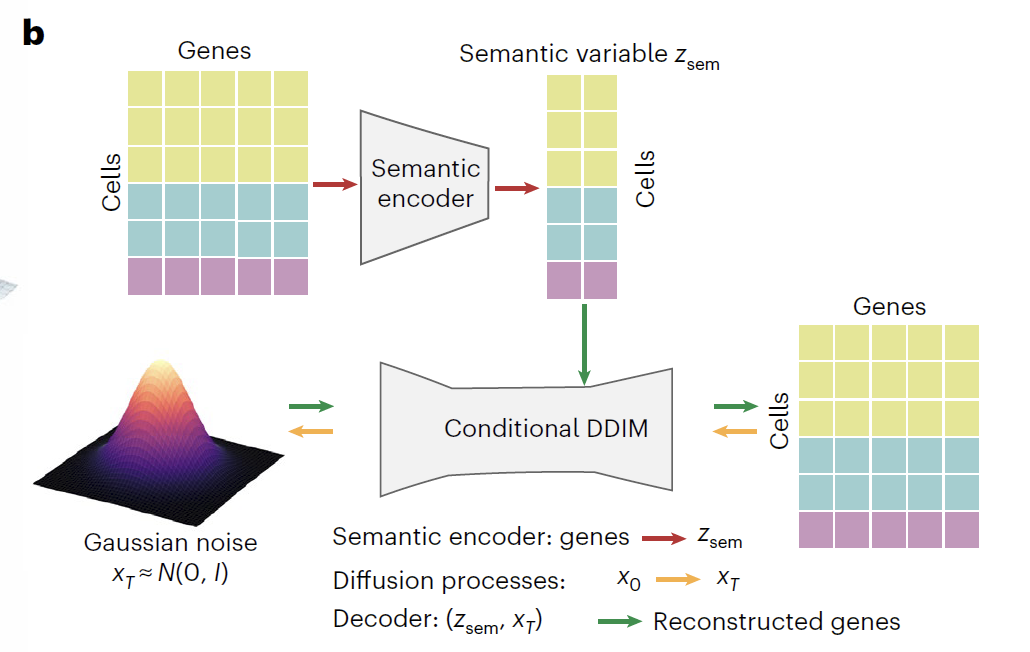
Das Herzstück von Squidiff bilden ein semantischer Encoder und ein Modul zur bedingten DDIM-Diffusion. Der semantische Encoder nutzt ein mehrschichtiges Perzeptron (MLP), um Einzelzell-RNA-Sequenzierungsdaten in einen niedrigdimensionalen semantischen Raum abzubilden und semantische Variablen (Z_sem) zu generieren, die Informationen zum Zelltyp und zu Perturbationen enthalten. Für die Arzneimittelforschung integriert dieser Encoder rekalibrierte Fingerabdrücke funktioneller Klassen (r_FCFP), die die Molekülstruktur des Arzneimittels als 2048-dimensionalen Vektor im semantischen Raum kodieren. Um unbekannte Arzneimittelperturbationen vorherzusagen, enthält das Modell außerdem ein Adaptermodul, das die Eingabe von Arzneimittel-SMILES-Strings und Dosierungsinformationen ermöglicht und so eine umfassende Fusion biologischer und chemischer Informationen erzielt.
Das bedingte DDIM-Modul folgt einem Dual-Prozess-Design aus Vorwärtsdiffusion (Genraumdiffusion) und Rückwärtsdiffusion (Genraum-Rückwärtsdiffusion).Während des Vorwärtsdiffusionsprozesses werden die ursprünglichen Genexpressionsdaten (x₀) durch 1.000 Iterationen schrittweise in annähernd reines Rauschen (x₀) umgewandelt.Dabei nähern sich die drei typischen Zelltypen schrittweise einer Gaußverteilung an, während Z_sem die biologischen Variationen der Genexpression effektiv erfasst und unterschiedliche experimentelle Bedingungen im latenten Raum klar voneinander trennt. Während des Rückdiffusionsprozesses wird ein Rauschvorhersagenetzwerk mit sinusförmiger Positionseinbettung (ε) verwendet.Unter Verwendung des Zeitschritts (t) und Z_sem als duale Bedingungen wurde das biologisch signifikante Transkriptom aus x_T durch iteratives Entrauschen rekonstruiert, wodurch das ursprüngliche Transkriptomprofil erfolgreich wiederhergestellt wurde.
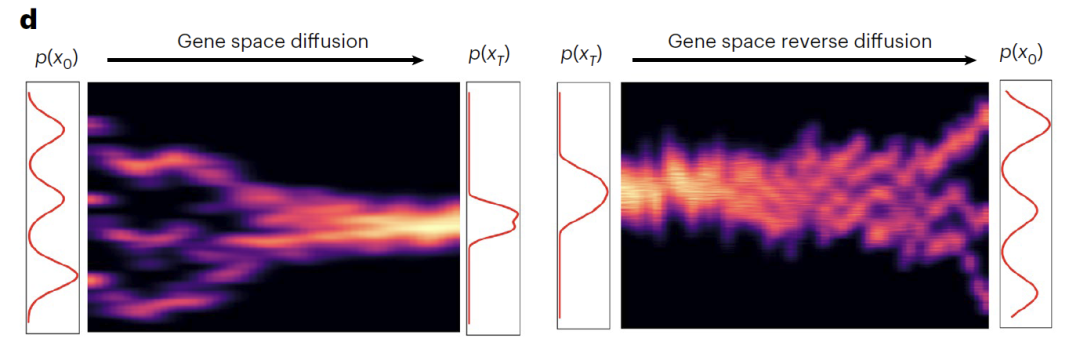
Das Modelltraining konzentriert sich auf den Rauschvorhersageverlust als zentrales Optimierungsziel, verwendet den Adam-Optimierer (Lernrate 1×10⁻⁴) und setzt auf GPU-Beschleunigung.Durch die Koordination der Regulierung von Zeitschritten und semantischen Variablen kann das Modell die kontinuierliche Entwicklung von Zellzuständen simulieren und so die Vorhersage dynamischer Trajektorien unterstützen.
Im Vergleich zu traditionellen Variations-Autoencodern bietet Squidiff erhebliche Vorteile:Ohne die Annahme einer Gaußverteilung zu erfordern, erfasst es komplexe Genexpressionsmuster durch feine Rauschunterdrückung und verbessert den F1-Score um 27% bei der Vorhersage seltener Zelltypen (<5%). Es führt innovativ die „Gradienteninterpolationsstrategie“ ein, die kontinuierliche Differenzierungspfade durch lineare Kombination semantischer Variablen im latenten Raum generiert und erfolgreich transiente Zellzustände identifiziert, die von traditionellen Modellen leicht übersehen werden (wie z. B. Mesodermvorläuferzellen bei der iPSC-Differenzierung).
Darüber hinaus bietet das Modell zwei Methoden zur Manipulation latenter Variablen: Die „Addition“ kombiniert die ursprüngliche Repräsentation mit der Störungsrichtung (Δz_sem), wie in Abbildung f unten dargestellt, um die Genexpressionsverteilung zu verschieben und den Störungseffekt widerzuspiegeln; die „Interpolation“ verwendet lineare Interpolation, wie in Abbildung g unten dargestellt, um kontinuierliche Zustände zu erzeugen, indem Zwischenpunkte auf der Vektorverbindungslinie ermittelt werden, wodurch ein sanfter Übergang zwischen Zelltypen erreicht wird.
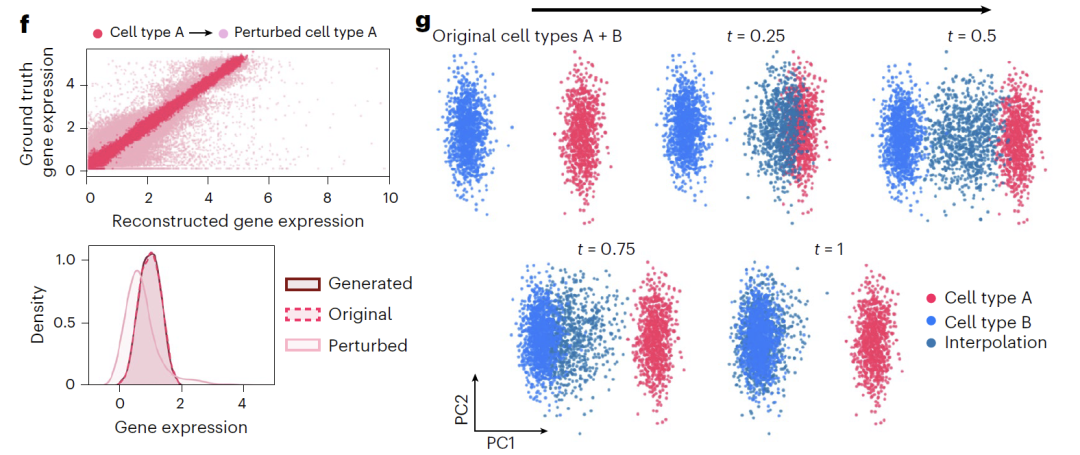
Squidiff-Multi-Szenario-Demonstration: Präzise Erfassung von transkriptomischen Veränderungen bei Zelldifferenzierung, Störungen und Strahlenreaktion
Um die Vorhersagefähigkeiten von Squidiff für das Transkriptom systematisch zu überprüfen, führte das Forschungsteam experimentelle Untersuchungen in vier Schlüsselbereichen durch: Zelldifferenzierung, Gen- und Arzneimittelperturbation, Entwicklung vaskulärer Organoide und Strahlenschäden.
Wie in der Abbildung unten dargestellt, trainierte das Team das Modell zur Vorhersage der Zelldifferenzierung ausschließlich mit Daten von Tag 0 und Tag 3 des iPSC-zu-Endoderm-Differenzierungsdatensatzes. Die Differenzierungsrichtung wurde durch die Berechnung semantischer Variablendifferenzen ermittelt, und Squidiff sagte den Zwischenzustand zwischen Tag 1 und 2 erfolgreich voraus. Das Modell erfasste präzise die Herunterregulierung des Pluripotenzmarkers MMOG, die Hochregulierung des Endodermfaktors GATA6 und identifizierte die transiente Expression des mesodermalen Markers DBX1. Im Vergleich zu traditionellen Methoden ermöglicht das von Squidiff generierte Transkriptom die Rekonstruktion eines kontinuierlichen Entwicklungspfads, der sehr gut mit der tatsächlichen Entwicklungstrajektorie übereinstimmt.
Das Modell zeigt eine hervorragende Leistung bei der Vorhersage von Gen- und Arzneimittelveränderungen.Bei Dual-Gen-Knockout-Experimenten in K562-Zellen kann Squidiff nicht-additive Effekte ohne Vorwissen präzise vorhersagen, und seine Robustheit übertrifft die bestehender Methoden.In klinischen Studien konnte das Modell die synergistischen Effekte von Kombinationspräparaten allein anhand von Einzelwirkstoffdaten vorhersagen und die spezifischen Wirkungen von Pabicept auf Tumorzellen präzise identifizieren. Durch die Integration eines Wirkstoffadapters erreichte das Modell zudem für den unbekannten Wirkstoff SGLT1 eine mit spezialisierten Modellen vergleichbare Vorhersagegenauigkeit und demonstrierte damit seine ausgezeichnete Generalisierungsfähigkeit.
In ihrer Forschung zu vaskulären Organoiden (BVO) konnte das Team mithilfe eines iPSC-induzierten BVO-Modells erfolgreich Zellzustände zu mehreren Zwischenzeitpunkten vorhersagen.Das Modell reproduzierte nicht nur die Differenzierungswege der drei Hauptzelltypen – Endothelzellen, Fibroblasten und Wandzellen –, sondern identifizierte auch das Zwischenstadium der Wandzelldifferenzierung zu Endothelzellen, das mit traditionellen Methoden schwer zu erfassen ist. Die Genexpressionsanalyse zeigte, dass die charakteristischen Genveränderungen in den vorhergesagten Daten weitgehend mit bekannten Entwicklungsmustern übereinstimmten.
In Studien zu Strahlenschäden sagte das Modell die Auswirkungen von Strahlung auf verschiedene Zelltypen präzise voraus, wobei lediglich Trainingsdaten von Endothelzellen verwendet wurden. Die Analyse zeigte, dass sich Zellen im frühen Entwicklungsstadium empfindlicher gegenüber Strahlung zeigten. Die vom Modell vorhergesagten differentiell exprimierten Gene und zugehörigen Signalwege wurden experimentell bestätigt. Bei der Vorhersage der Schutzwirkung von G-CSF deckte das Modell die Schutzmechanismen des Medikaments gegen verschiedene Zelltypen auf: Aktivierung von Angiogenese-Signalwegen in Fibroblasten, Hemmung von Apoptose-Signalwegen in Endothelzellen und erhöhte genomische Stabilität in Wandzellen. Die experimentelle Validierung zeigte, dass der Zelltod nach der G-CSF-Behandlung signifikant reduziert war, was die Zuverlässigkeit der Modellvorhersagen belegt.
Diese Systemsexperimente zeigen, dass Squidiff nicht nur Veränderungen des Zellzustands unter verschiedenen biologischen Szenarien präzise vorhersagen kann, sondern auch transiente Zustände erfasst und unbekannte Störungen ableitet. Damit bietet es ein leistungsstarkes und zuverlässiges Computerwerkzeug zur Vorhersage von Zellreaktionen.
KI-gestütztes neues Paradigma für die Einzelzellforschung
Im interdisziplinären Feld der Einzelzellbiologie und der künstlichen Intelligenz treibt der von Squidiff repräsentierte Durchbruch in der Diffusionsmodelltechnologie die kollaborative Innovation zwischen Wissenschaft und Industrie voran.
Auf akademischer Forschungsebene erzielen führende Universitätsteams weltweit weiterhin bahnbrechende Fortschritte in der Tiefe und Breite der Einzelzellmodellierung.Ein Forschungsteam der Universität Toronto in Kanada hat scGPT entwickelt und veröffentlicht, das erste grundlegende groß angelegte Sprachmodell für die Einzelzellbiologie.Das Modell basiert auf einer generativen, vortrainierten Transformer-Architektur und wurde mit über 33 Millionen Zelldatenpunkten trainiert, die 51 menschliche Organe/Gewebe und 441 unabhängige Studien umfassen. Es deckt umfassend verschiedene Zelltypen sowie physiologische und pathologische Zustände ab und stellt einen detaillierten Atlas der menschlichen Zellheterogenität dar.
Titel des Artikels:scGPT: Auf dem Weg zu einem grundlegenden Modell für die Einzelzell-Multi-Omik mithilfe generativer KI
Papieradresse:
https://biorxiv.org/content/10.1101/2023.04.30.538439
gleichzeitig,Das Team der Stanford University konzentrierte sich auf Innovationen in der räumlichen Dimension und entwickelte das dreidimensionale raumzeitliche Modellierungsframework Spateo.Auf Basis skalierbarer und präziser Algorithmen kann dieses Framework vollständige dreidimensionale Embryo- und Organmodelle aus kontinuierlichen zweidimensionalen Gewebeschnittdaten rekonstruieren und ein mehrstufiges räumliches digitales System aufbauen, das von molekularen Einzelzellmerkmalen bis zur makroskopischen Embryomorphologie reicht.
Titel des Artikels:Räumlich-zeitliche Modellierung von Molekülhologrammen
Papieradresse:
https://www.cell.com/cell/fulltext/S0092-8674(24)01159-0
Die Wirtschaft setzt diese akademischen Erkenntnisse in praktische Werkzeuge um und demonstriert damit ihren bedeutenden Wert bei der Arzneimittelentwicklung, der Behandlung von Krankheiten und in anderen Anwendungsbereichen.Cell2Sentence-Scale 27B (C2S-Scale 27B), entwickelt von Google in Zusammenarbeit mit der Yale University und anderen Institutionen, ist eines der weltweit größten Basismodelle für die Einzelzellanalyse.Dieses auf der Open-Source-Modellfamilie Gemma basierende Modell verfügt über 27 Milliarden Parameter und ermöglicht die detaillierte Analyse von Genexpressionsmustern in einzelnen Zellen sowie die präzise Vorhersage zellulärer Reaktionen auf medikamentöse Interventionen. Aktuell ist das Modell in die Wirkstoff-Screening-Plattform von Google Health integriert und unterstützt die Entwicklung personalisierter Kombinationstherapien für „kalte Tumore“ sowie die Beschleunigung der Entwicklung von Immuntherapien.Eine weitere wichtige Praxis ergibt sich aus der Zusammenarbeit zwischen dem gemeinnützigen Arc Institute und Unternehmen wie 10x Genomics, deren STATE-Modell sich auf die Simulation dynamischer zellulärer Reaktionen konzentriert.Es integriert Beobachtungsdaten von 170 Millionen Zellen und Interventionsdaten von 100 Millionen Zellen und ermöglicht so eine präzise Simulation von transkriptomischen Veränderungen in Zellen unter medikamentöser Behandlung, Genomeditierung oder Strahlenexposition.
Es ist nicht schwer zu erkennen, dass von der eingehenden Erforschung grundlegender Einzelzellmodelle durch die akademische Gemeinschaft bis hin zur großflächigen Implementierung der Technologie in der Industrie alles darauf zurückzuführen ist.Die Diffusionsmodellierungstechnologie von Squidiff treibt die Einzelzellforschung von der „Analyse des Zellzustands“ hin zur „Vorhersage des Zellschicksals“ voran.Dieser Sprung beschleunigt nicht nur den Fortschritt in Bereichen wie der Arzneimittelentwicklung und der Krebsbehandlung, sondern bietet auch eine grundlegende technologische Unterstützung für zukünftige medizinische Richtungen wie die Präzisionsmedizin und die regenerative Medizin und entfesselt kontinuierlich das enorme Potenzial KI-gesteuerter Innovationen in den Lebenswissenschaften.
Referenzartikel:
1.https://mp.weixin.qq.com/s/yCR_GC0Ln80st2tHcv08-Q
2.https://mp.weixin.qq.com/s/GegQB65w4nZG6ZXvnyU9dw








