Command Palette
Search for a command to run...
Mit Einer Genauigkeit Von 971 TP3T! Die Princeton University Und Andere Schlugen MOFSeq-LMM Vor, Das Effizient Vorhersagt, Ob MOFs Synthetisiert Werden können.
Metallorganische Gerüstverbindungen (MOFs) haben aufgrund ihrer hochgradig einstellbaren Porenstrukturen und vielfältigen chemischen Funktionalitäten großes Potenzial für Anwendungen wie Gasspeicherung, -trennung, Katalyse und Wirkstofffreisetzung gezeigt.MOFs verfügen über einen riesigen Gestaltungsraum mit Billionen möglicher Kombinationen von Bausteinen, was die alleinige experimentelle Erkundung extrem ineffizient macht.
Um die Entdeckung von MOFs zu beschleunigen, wurden computergestützte Verfahren entwickelt, die darauf abzielen, neuartige MOFs zu generieren, ihre Eigenschaften vorherzusagen und letztendlich ihre Synthese zu erreichen. In diesem ProzessDie größte Herausforderung besteht in der geringen Konversionsrate vom Screening zur Synthese.Dies rührt größtenteils von der Unsicherheit hinsichtlich der Machbarkeit der computergenerierten Synthese von MOFs her. Beispielsweise wurden von den Tausenden bisher veröffentlichten computergestützten MOF-Screenings nur etwa ein Dutzend durch eine MOF-Synthese begleitet.
Die freie Energie ist ein wichtiger Indikator zur Bewertung der thermodynamischen Stabilität und Synthetisierbarkeit von MOFs. Herkömmliche Rechenmethoden sind jedoch bei großen MOF-Datensätzen rechenintensiv, was ein schnelles Screening erschwert. Um diese Herausforderung zu bewältigen, hat ein gemeinsames Forschungsteam der Princeton University und der Colorado School of Mines eine effiziente, auf maschinellem Lernen basierende Vorhersagemethode entwickelt.Durch die Verwendung großer Sprachmodelle (LLM) zur direkten Vorhersage der freien Energie aus der Struktursequenz von MOFs können die Rechenkosten erheblich reduziert werden, was eine Hochdurchsatz- und skalierbare thermodynamische Bewertung von MOFs ermöglicht.Das Modell zeichnet sich durch hohe Vielseitigkeit aus, ohne dass ein erneutes Training erforderlich ist: Sein F1-Wert liegt bei 97%, wenn es darum geht, festzustellen, ob die freie Energie von MOFs höher oder niedriger als die empirisch ermittelte synthetische Machbarkeitsschwelle ist.
Die zugehörigen Forschungsergebnisse mit dem Titel „Hochpräzise und schnelle Vorhersage der freien Energie von MOFs mittels maschinellen Lernens“ wurden in ACS Publications veröffentlicht.
Forschungshighlights:
Auf der Grundlage dieses Modells können Forscher die freie Energie mit hoher Genauigkeit vorhersagen und die Ergebnisse vollständiger Molekülsimulationen ohne erneutes Training simulieren, wodurch die Machbarkeit der Synthese von MOFs bestimmt wird.
* Arbeiten, die früher viel Zeit im Labor oder durch Molekülsimulationen erforderten, können jetzt in vernachlässigbarer Zeit erledigt werden.
Diese Methode bietet einen praktikablen Ansatz für die Verwendung von maschinellem Lernen zur Vorhersage der freien Energie als frühes oder spätes Screening-Instrument beim leistungsbasierten computergestützten MOF-Screening.

Papieradresse:
https://pubs.acs.org/doi/10.1021/jacs.5c13960
Folgen Sie unserem offiziellen WeChat-Konto und antworten Sie im Hintergrund mit „free energy prediction“, um das vollständige PDF zu erhalten.
Weitere Artikel zu den Grenzen der KI:
MOFMinE: Eine Million MOF-Prototypen im Fokus
Zur Unterstützung des ModelltrainingsDas Forschungsteam erstellte einen riesigen MOF-Datensatz, MOFMinE, der ungefähr eine Million MOF-Prototypen umfasst.Sie umfasst Informationen zum gesamten Prozess von der Komponentenauswahl und der Zuordnung von Topologievorlagen bis hin zu funktionalen Modifikationen, wie in der folgenden Abbildung dargestellt:
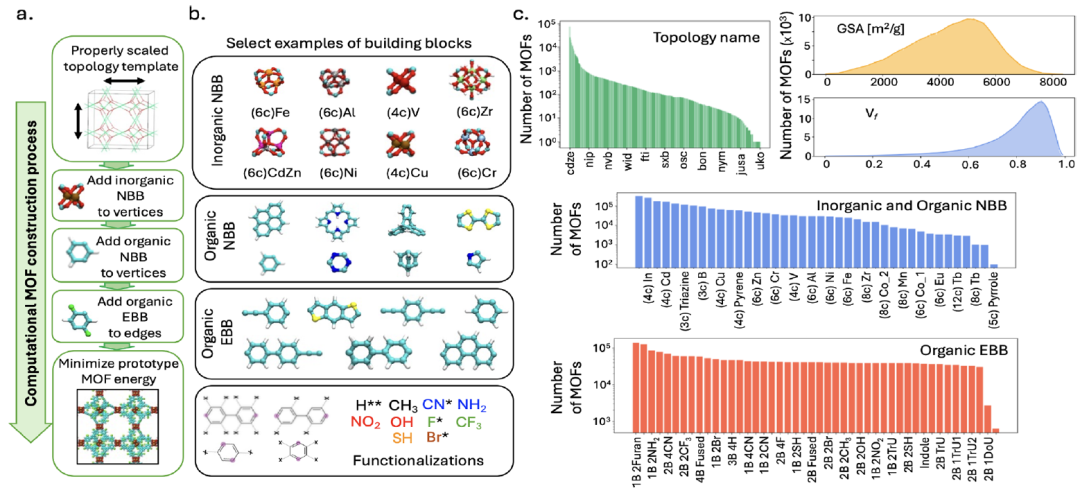
Konstruktionsmethode
Der Datensatz basiert auf der ToBaCCo-3.0-Plattform. Jedes MOF wird generiert, indem seine Bausteine auf eine topologische Vorlage abgebildet werden, die entsprechend der Größe des Bausteins skaliert wurde. Diese Vorlage steuert die räumliche Anordnung und Vernetzung der Bausteine innerhalb der MOF-Zelle. ToBaCCo-Bausteine werden anhand ihrer Position in Knotenbausteine (NBBs) und Kantenbausteine (EBBs) unterteilt: Knotenbausteine werden auf Eckpunkte der Vorlage abgebildet, Kantenbausteine auf Kanten. NBBs können anorganisch oder organisch sein. Anorganische NBBs entsprechen den sogenannten MOF-Bausteinen zweiter Ordnung (SBUs), während organische NBBs sich mit EBBs zu MOF-Konnektoren verbinden.
Datenumfang und -diversität
MOFMinE enthält 1.393 topologische Vorlagen, 27 anorganische NBBs, 14 organische NBBs und 19 grundlegende EBBs und umfasst 13 funktionelle Modifikationen, wodurch eine Vielfalt an chemischen und topologischen Strukturen gewährleistet wird.Die Datenbank weist einen Hohlraumanteil von 0,01 bis 0,99, eine Oberfläche (GSA) von 26 bis 8382 m²/g und eine maximale Porengröße (LPD) von 2,6 bis 127,7 Å auf und deckt damit den gesamten Strukturraum von MOFs ab.
Teilmenge der freien Energie
Von diesen 1 Million MOF-Prototypen wurden für eine Untergruppe von 65.574 Strukturen Daten zur freien Energie erhoben.Diese Teilmenge enthält 379 topologische Vorlagen, 6 anorganische NBBs, 11 organische NBBs und 12 basische EBBs mit 13 Funktionalisierungsmodifikationen. Die Porositätseigenschaften der Teilmenge sind: Vf von 0,01 bis 0,97, GSA von 38 bis 7304 m²/g und LPD von 2,6 bis 87,8 Å. Dieser Datensatz wurde zur Feinabstimmung der freien Energievorhersage und zum Testen von LLMs verwendet.
MOFSeq-LMM-Modell zur effizienten Vorhersage der freien Energie von MOFs
Mit Unterstützung des MOFMinE-Datensatzes,Das Forschungsteam entwickelte das MOFSeq-LMM-Modellframework, um die freie Energie von MOFs effizient vorherzusagen und ein datengetriebenes Design von der Struktur bis zu den Eigenschaften zu realisieren.Die Kernidee dieses Frameworks besteht darin, die Strukturinformationen von MOFs in eine computerverständliche Sequenzdarstellung (MOFSeq) umzuwandeln und diese mit einem großen Sprachmodell zum Lernen und Vorhersagen zu kombinieren, wodurch die Rechenkosten erheblich reduziert werden, während gleichzeitig die physikochemischen Informationen erhalten bleiben.
MOFSeq-Charakterisierung
Um die Einschränkungen bestehender Repräsentationsstrategien zu überwinden und große Sprachmodelle für die umfassende Vorhersage von MOF-Eigenschaften voll auszuschöpfen,Forscher haben MOFSeq entwickelt. Diese neuartige, auf Zeichenketten basierende Sequenzdarstellungsmethode ist sowohl kompakt als auch hoch informativ und kodiert die lokalen und globalen Strukturmerkmale von MOFs auf optimierte Weise.Dies ermöglicht die effiziente und skalierbare Verarbeitung von Sprachmodellen.
In MOFSeq umfassen lokale Informationen hauptsächlich die atomare Zusammensetzung der Bausteine und deren interne Vernetzung; globale Informationen umfassen hauptsächlich übergeordnete Beschreibungen der MOF-Bausteine und die Verbindungsmuster zwischen den Bausteinen. Lokale Informationen werden mithilfe des MOFid-Tools gewonnen, während globale Informationen auf ToBaCCo-3.0 basieren, wie in der folgenden Abbildung dargestellt:
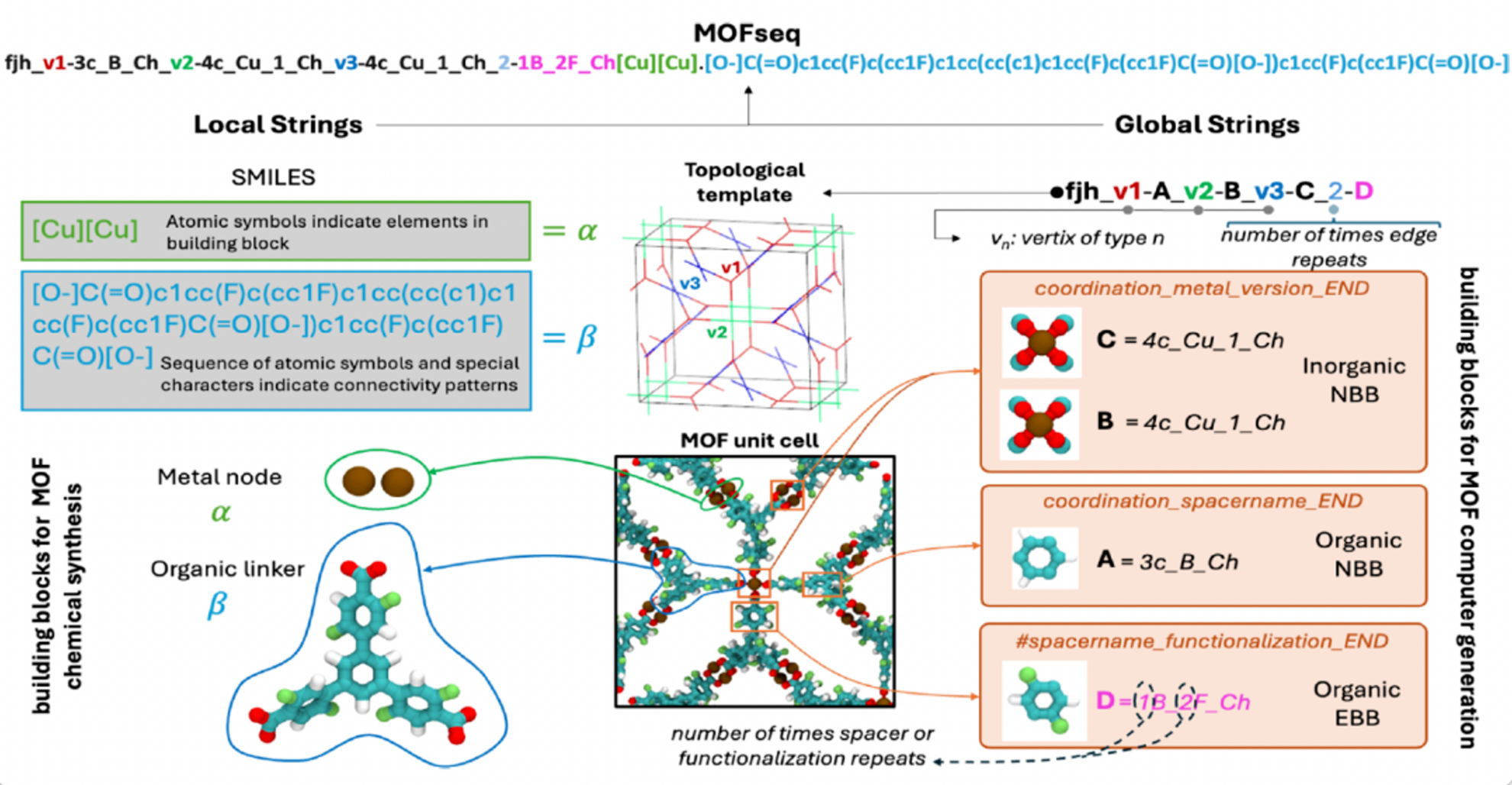
MOFs-Datenbankaufbau und Datenverarbeitung
Nach der Erstellung des MOFMinE-Datensatzes auf Basis der oben beschriebenen Methode wurden alle von ToBaCCo generierten MOF-Prototypen unter Verwendung des UFF4MOF-Kraftfelds in LAMMPS (Version vom 29. Oktober 2020) optimiert, um die endgültigen MOF-Strukturen zu erhalten.
Der mit ToBaCCo-3.0 generierte Datensatz enthält lediglich den MOF-Namen und die zugehörige CIF-Datei als Repräsentation jedes MOF. MOFSeq benötigt jedoch sowohl den MOF-Namen als auch die MOFid.Um MOFid zu erhalten, verwendeten die Forscher den von Bucior et al. entwickelten MOFid-Generator.Dieser Generator kann MOFid und MOFkey gleichzeitig auf Basis der CIF-Struktur von MOF generieren.
Die 793.079 MOFSeq-Vortrainingsproben wurden schließlich in einen Trainingsdatensatz mit 634.463 Proben, einen Validierungsdatensatz mit 79.308 Proben und einen Testdatensatz mit 79.308 Proben aufgeteilt. Die 54.443 MOFSeq-Feinabstimmungsdatenpunkte wurden in einen Trainingsdatensatz mit 43.554 Proben, einen Validierungsdatensatz mit 5.444 Proben und einen Testdatensatz mit 5.445 Proben aufgeteilt.
LLM-Prop-Modelldesign
Aufbauend auf der MOFSeq-Charakterisierung setzte das Forschungsteam LLM-Prop ein, ein großes Sprachmodell, das speziell für die Vorhersage von Materialeigenschaften entwickelt wurde. Das LLM-Prop-Modell ist mit etwa 35 Millionen Parametern relativ klein und gewährleistet so sowohl Lernfähigkeit als auch Recheneffizienz. Die Eingabelänge des Modells ist auf 2.000 Token festgelegt, wodurch die Struktursequenzinformationen der meisten MOFs abgebildet werden können. Mithilfe eines Aufmerksamkeitsmechanismus erfasst das Modell adaptiv den Einfluss verschiedener Komponenten und Topologien auf die freie Energie in der Sequenz und bildet so eine interaktive Repräsentation globaler und lokaler Merkmale.
Vortraining und Feinabstimmung
* Vorbereitungsphase:
Forscher trainierten LLM-Prop, um die Dehnungsenergien von MOFs mithilfe der MOFSeq-Repräsentation vorherzusagen. Die Dehnungsenergie wurde aufgrund ihres geringen Rechenaufwands und ihrer hohen Korrelation mit der freien Energie gewählt. Während des Vortrainings wurden Dropout-Raten von 0,2 und 0,5 verwendet. Die Ergebnisse zeigten, dass eine Dropout-Rate von 0,2 sowohl im Vortraining als auch in den nachfolgenden Aufgaben bessere Ergebnisse lieferte. Die Eingabelänge von MOFSeq wurde auf 2000 Token festgelegt.
Feinabstimmungsphase:
Die Einrichtung entspricht der Vorab-Trainingsmethode, jedoch wurde das Modellziel auf die Vorhersage der freien Energie geändert und die Anzahl der Trainingsepochen auf 200 erhöht. LLM-Prop ist als leichtgewichtiges Modell konzipiert und etwa 1/2000 so groß wie Llama-2, wobei die Recheneffizienz im Vordergrund steht. Dieser Ansatz birgt einen Kompromiss: Im Vergleich zum Feinabstimmen großer LLMs (wie Llama-2 oder GPT-2) benötigt LLM-Prop mehr Trainingsepochen, um eine hohe Leistung zu erzielen, seine geringe Größe ermöglicht jedoch ein praktikables und effizientes Training.
Die Genauigkeit der Vorhersage der MOF-Synthese erreichte 97%.
Nach dem Training des MOFSeq-LMM-Modells evaluierte das Forschungsteam systematisch dessen Leistungsfähigkeit bei der Vorhersage der freien Energie, der Bewertung der Synthesemachbarkeit und dem Screening polymorpher MOFs. Die experimentellen Ergebnisse bestätigten nicht nur die hohe Genauigkeit des Modells, sondern unterstrichen auch sein Anwendungspotenzial für das Hochdurchsatz-Design und -Screening von MOFs.
Vorhersageleistung der freien Energie
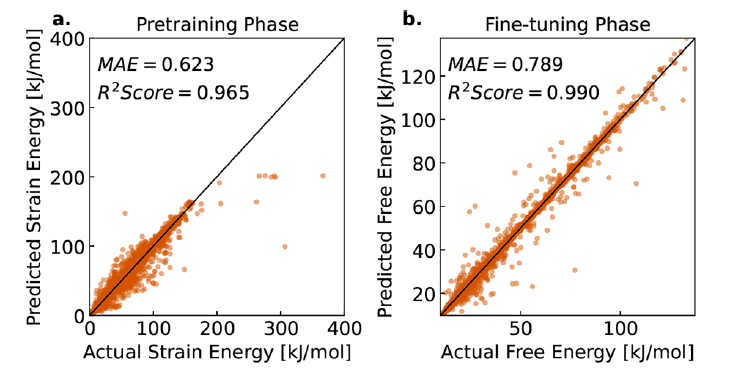
Erste,Das Team bewertete die Vorhersagegenauigkeit der freien Energie von LLM-Prop an unbekannten MOF-Proben.Die Ergebnisse zeigen, dass das Modell die freie Energie mit einem mittleren absoluten Fehler (MAE) von 0,789 kJ/mol MOFatom genau vorhersagen kann und dabei eine hohe Korrelation von R² = 0,990 erreicht, wie in Abbildung b unten dargestellt.Dies bedeutet, dass das Modell in der überwiegenden Mehrheit der MOF-Proben Vorhersagen liefern kann, die nahe an den wahren Werten liegen.
In der Vortrainingsphase wurde das Modell anhand von Dehnungsenergiedaten trainiert. Dabei wurde ein mittlerer absoluter Fehler (MAE) von 0,623 kJ/mol MOFatom und ein Bestimmtheitsmaß (R²) von 0,965 erreicht (siehe Abbildung a). Diese hohe Korrelation zeigt, dass Dehnungsenergiedaten effektive Vorabinformationen für die Vorhersage der freien Energie liefern können und bestätigt somit die Rationalität der Vortrainingsstrategie des Forschungsteams. Weiterführende Analysen belegen eine hohe Korrelation zwischen der vortrainierten Dehnungsenergie und der feinabgestimmten freien Energie. Dies unterstreicht den Wert der Dehnungsenergie als kostengünstigen Ersatzindikator im Modelltraining.
Ergebnisse des Ablationsexperiments
Um die Ursachen der Modellleistung besser zu verstehen, führte das Team systematische Ablationsexperimente durch. Die Experimente untersuchten den Einfluss lokaler und globaler Merkmale sowie des Vortrainings auf die Vorhersage der freien Energie. Die Ergebnisse sind in der folgenden Tabelle dargestellt:
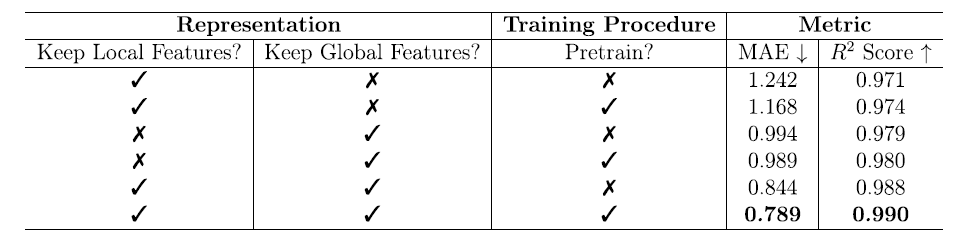
Nur lokale Merkmale: Durch das Vortraining sank der MAE von 1,242 auf 1,168 kJ/mol MOFatom, und R² stieg von 0,971 auf 0,974, was darauf hindeutet, dass das Vortraining die Generalisierungsfähigkeit des Modells verbessern kann, wenn nur wenige lokale Merkmale vorliegen.
* Nur globale Funktionen:
Die Leistung ist deutlich besser als bei der Verwendung ausschließlich lokaler Merkmale: Der MAE sinkt auf unter 1,0 kJ/mol MOFatom und der R²-Wert steigt auf etwa 0,980. Das Vortraining hat in diesem Fall einen relativ geringen Einfluss (der MAE sinkt von 0,994 auf 0,989 kJ/mol MOFatom und der R²-Wert steigt von 0,979 auf 0,980), was darauf hindeutet, dass globale Merkmale selbst mehr Aufgabeninformationen enthalten und weniger auf Vortraining angewiesen sind, um effektives Lernen zu ermöglichen.
* Kombination lokaler und globaler Merkmale:
Mit Unterstützung des Vortrainings erreichte das Modell eine optimale Leistung mit einem MAE von 0,789 kJ/mol MOFatom und einem R² von 0,990, was zeigt, dass der Synergieeffekt der beiden Merkmalsarten entscheidend für die Verbesserung der Vorhersagegenauigkeit ist.
Dieses Ablationsexperiment zeigt deutlich, dass die Gestaltung globaler und lokaler Merkmale sowie die Vortrainingsstrategie von MOFSeq die Kernelemente für die Verbesserung der Vorhersagefähigkeit des Modells sind.
Machbarkeitsbewertung der Synthese
In industriellen Anwendungen ist es wichtiger, die synthetische Machbarkeit von MOFs zu bestimmen, als sich lediglich auf den Absolutwert der freien Energie zu konzentrieren. Das Forschungsteam setzte ΔL_MFFL (einen auf der Korrektur der freien Energie basierenden Index) auf einen Schwellenwert von 4,4 kJ/mol MOF-Atom und führte eine binäre Klassifizierung zur Vorhersage der synthetischen Machbarkeit von MOFs durch. Die experimentellen Ergebnisse sind in der folgenden Abbildung dargestellt:
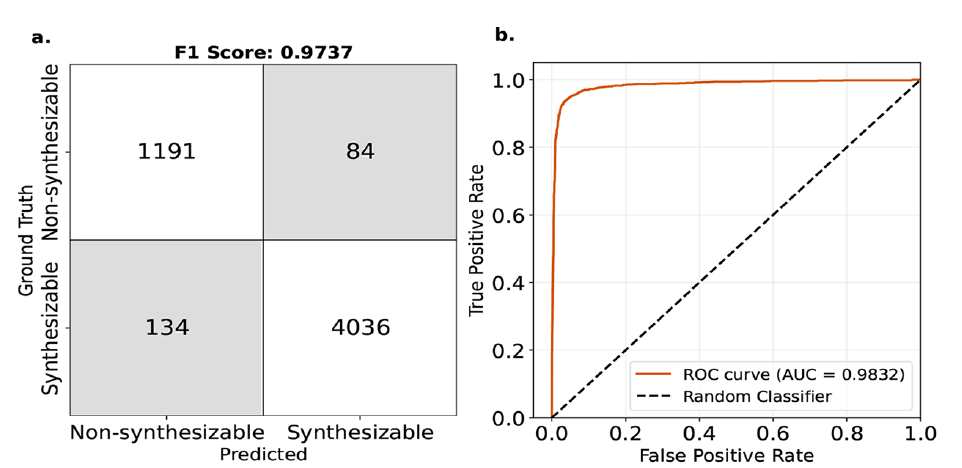
* Der F1-Score von 97% belegt die gute Generalisierungsfähigkeit des Modells.
* Die Fläche unter der ROC-Kurve (AUC) beträgt bis zu 0,98 – was letztendlich als die Wahrscheinlichkeit einer falschen Einschätzung verstanden werden kann, wenn das Modell feststellt, dass ein bestimmtes MOF synthetisierbar ist, die nur etwa 2% beträgt.
Screening polymorpher MOFs
Für MOF-Systeme mit Polymorphismus,Das Experiment bestätigte zudem die Fähigkeit des Modells, die stabilste polymorphe Form zu identifizieren.Unter 7.490 Polymorphfamilien, die jeweils 2–50 Polymorphe enthalten, kann das Modell den stabilsten Polymorph mit einer freien Energiedifferenz von nur 0,16 kJ/mol MOFatom korrekt auswählen, mit einer Erfolgsquote von etwa 63%; wenn die freie Energiedifferenz auf 0,49 kJ/mol MOFatom ansteigt, erhöht sich die Erfolgsquote auf 89%.
Insgesamt erreichte das Modell bei der Polymorphismuserkennung eine durchschnittliche Erfolgsquote von etwa 781 TP3T.Wie aus der folgenden Abbildung hervorgeht, besitzt es einen signifikanten Wert für die Hochdurchsatzvorhersage vor dem experimentellen Screening.

Aus praktischer Sicht liegt die Wahrscheinlichkeit für die korrekte Synthese eines bestimmten MOF-Designs durch LLM unter Berücksichtigung der thermodynamischen Stabilität und der polymorphen Konkurrenz zwischen etwa 76% und 98%. Die höhere Wahrscheinlichkeit entspricht dem Fall, dass das MOF keine konkurrierenden Polymorphe aufweist.
Künstliche Intelligenz verändert das Paradigma der MOF- und Materialwissenschaftsforschung.
8. Oktober 2025Die Königlich Schwedische Akademie der Wissenschaften hat beschlossen, den Nobelpreis für Chemie 2025 an Professor Susumu Kitagawa von der Universität Kyoto, Professor Richard Robson von der Universität Melbourne und Professor Omar Yaghi von der Universität von Kalifornien, Berkeley, zu verleihen, in Anerkennung ihrer Forschungsbeiträge auf dem Gebiet der MOFs.Im Rückblick auf diesen historischen Moment hat die MOF-Forschung eine über 30-jährige Entwicklung durchlaufen, die sich schrittweise von der anfänglichen Strukturierung und Syntheseforschung hin zur Leistungsregulierung, Anwendungserweiterung und Industrialisierung entwickelt hat. Nach diesem Meilenstein begrüßt die Materialwissenschaft eine neue Variable: Die tiefgreifende Einbindung künstlicher Intelligenz verändert das Forschungsparadigma und den Innovationsrhythmus von MOFs und sogar des gesamten Feldes der Materialwissenschaft.
Als Antwort auf die Herausforderung einer riesigen, komplexen Welt von MOFs ohne standardisierte Namenskonventionen, im Oktober 2025Ein Forschungsteam der Universität Toronto und des Clean Energy Innovation Research Centre des National Research Council of Canada hat MOF-ChemUnity vorgeschlagen: einen strukturierten, skalierbaren und erweiterbaren Wissensgraphen.Diese Methode nutzt LLM, um eine zuverlässige Eins-zu-eins-Zuordnung zwischen MOF-Namen und ihren Synonymen in der Literatur sowie in der CSD registrierten Kristallstrukturen herzustellen und so eine eindeutige Unterscheidung zwischen MOF-Namen, ihren Synonymen und Kristallstrukturen zu ermöglichen. In der aktuellen Version integriert MOF-ChemUnity ca. 10.000 wissenschaftliche Artikel und über 15.000 CSD-Kristallstrukturen sowie deren berechnete chemische Eigenschaften in einem maschinenlesbaren Format.
Titel des Papers: MOF-ChemUnity: Literaturbasierte große Sprachmodelle für die Forschung an metallorganischen Gerüstverbindungen
Papieradresse:https://pubs.acs.org/doi/10.1021/jacs.5c11789
Bei der rationalen Entwicklung von MOF-Materialien stellt die Vorhersage der Struktur vor der Synthese seit jeher eine zentrale Herausforderung für die effiziente und gezielte Synthese dieser Materialien dar. Um dieses Problem zu lösen,Ein Team unter der Leitung der Professoren Cui Yong und Gong Wei an der Shanghai Jiao Tong Universität hat einen datengesteuerten maschinellen Lernprozess entwickelt, der eine schnelle und genaue Vorhersage des Metallknotentyps von MOFs ermöglicht.Diese Methode nutzt die Strukturinformationen organischer Liganden als Eingangsdaten und stellt mithilfe eines maschinellen Lernmodells eine Zuordnung zwischen Ligandeneigenschaften und Metallknotentypen her. Dadurch lassen sich die potenziell entstehenden Metallknotentypen vor der Synthese effektiv vorhersagen. Das trainierte und optimierte Vorhersagemodell erreichte im Testdatensatz eine Genauigkeit von 91%, eine Präzision von 89% und einen Recall von 85%.
Titel der Arbeit: Datengetriebenes maschinelles Lernen zur Vorhersage von Metallknotentypen in metallorganischen Gerüsten zur Steuerung des Linkerdesigns und zur gezielten inversen C3H8/C3H6-Trennung
Papieradresse:http://engine.scichina.com/doi/10.1007/s11426-025-2917-4
Die traditionelle MOF-Forschung beginnt oft mit der Struktur oder den Eigenschaften und nähert sich dem Zielmaterial schrittweise durch lokale Variablenkontrolle und umfangreiche Experimente oder Berechnungen an. In diesen neuen Arbeiten verschiebt sich jedoch der Ausgangspunkt selbst: Forscher entwickeln zunächst rechnerisch realisierbare und plausible Materialdarstellungssysteme und lassen die Modelle dann lernen, welche Strukturkombinationen physikalisch plausibel, thermodynamisch realisierbar und synthetisch sinnvoll sind. Wenn Modelle schnell zuverlässige thermodynamische und strukturelle Aussagen im Millionenbereich von Strukturen liefern können, verlagert sich der Fokus der Materialforschung nach oben – von der Frage „Wie berechnet und misst man?“ hin zur Frage „Wie definiert man das Problem, erstellt Darstellungen und legt Entscheidungsgrenzen fest?“. Dies könnte der nächste methodische Sprung sein, den die MOF-Forschung nach über dreißig Jahren struktureller und chemischer Erkenntnisse erreichen kann.
Quellen:
1.https://pubs.acs.org/doi/10.1021/jacs.5c13960
2.https://phys.org/news/2026-01-tool-narrows-ideal-metal-frameworks.html