Command Palette
Search for a command to run...
Von Quarz Bis Hin Zu Ferroelektrischen Materialien: Die Harvard University Schlägt Ein Äquivariantes Maschinelles Lernframework Vor, Um Die Groß Angelegte Elektrische Feldsimulation Von Materialien Zu Beschleunigen

Als wegweisende Forschungsrichtung in der modernen Materialwissenschaft verfolgt die computergestützte Materialwissenschaft die zentrale Aufgabe, die Mikrostruktur von Materialien zu analysieren und ihre makroskopischen Eigenschaften vorherzusagen. Basierend auf grundlegenden physikalischen Gesetzen wie der Quantenmechanik zielt diese Disziplin darauf ab, die experimentell messbaren Eigenschaften realer Materialien präzise vorherzusagen und so ein tiefes Verständnis der Reaktionsmechanismen von Materialien auf äußere Reize zu erlangen. Zu diesen Reaktionseigenschaften gehören lineare, nichtlineare und Kopplungseffekte.Es ist das Kernelement, das die Funktionsleistung von Dielektrika, Ferroelektrika, multiferroischen Materialien und piezoelektrischen Materialien bestimmt.
Derzeit ist die Methode der elektronischen Strukturbestimmung auf der Grundlage der Dichtefunktionaltheorie (DFT) ein wichtiges Mittel zur Untersuchung der Eigenschaften von Materialien.Da der Rechenaufwand jedoch exponentiell mit der Größe des Systems steigt, können damit nur Systeme im kleinen Maßstab verarbeitet werden.Dies hat die systematische Erforschung komplexer Materialsysteme stark eingeschränkt. In den letzten Jahren hat die Einführung maschineller Lernmethoden zu Durchbrüchen auf diesem Gebiet geführt. Durch die Erstellung datenbasierter Modelle hat sich großes Potenzial für die Vorhersage von Materialpolarisation, Bornscher Ladung, Polarisierbarkeit, Dielektrizitätskonstante, spektralen Eigenschaften usw. gezeigt und wurde erfolgreich auf Moleküle, flüssiges Wasser und feste Materialsysteme angewendet.
Die meisten bestehenden Methoden des maschinellen Lernens weisen jedoch noch Einschränkungen auf. Beispielsweise ist es bei unabhängig entwickelten Modellen schwierig, die strikte Umsetzung physikalischer Symmetrien und Erhaltungssätze sicherzustellen. Einige Einzelmodelllösungen, die versuchen, dielektrische Eigenschaften, Energie- und Kraftberechnungen zu integrieren, stoßen bei der Erweiterung auf reale Systeme mit periodischen Randbedingungen und mehrwertiger Polarisation auf Herausforderungen. Obwohl einige Studien potenzielle Energieoberflächen durch das Training atomarer Kräfte unter verschiedenen elektrischen Feldern konstruierten und indirekt Polarisationseigenschaften ableiteten, um die Schwierigkeiten beim Training mehrwertiger Polarisationsdaten zu umgehen,Allerdings basiert diese Methode auf impliziten Ableitungsberechnungen, die die Vorhersagegenauigkeit verringern können, und das Problem der hohen Kosten der groß angelegten DFT-Datenerfassung wurde nicht grundsätzlich gelöst.
Um die oben genannten Herausforderungen zu bewältigen, haben die Harvard University und Robert Bosch LLC, eine Tochtergesellschaft der deutschen Bosch-Gruppe in den USA, gemeinsam ein einheitliches differenzierbares Lernframework für elektrische Reaktionen entwickelt.Dieses Framework kann gleichzeitig die verallgemeinerte potenzielle Energie und ihre Reaktionsfunktion auf externe Reize in einem einzigen maschinellen Lernmodell erlernen.Durch die Definition der Antwortfunktion als Ableitung der verallgemeinerten potentiellen Energie in Bezug auf die Atomkoordinaten und Störungsparameter, die Nutzung der präzisen mathematischen Beziehung zwischen beiden und die strikte Einhaltung physikalischer Beschränkungen wie Impulserhaltung und der Bornschen Summationsregel für akustische Ladungen werden die inhärenten Mängel traditioneller unabhängiger Modelle überwunden und ein neuer Weg für hochpräzise Forschungen zu den dielektrischen und ferroelektrischen Eigenschaften kristalliner, ungeordneter und flüssiger Materialien eröffnet.
Die entsprechenden Forschungsergebnisse wurden in der international renommierten Fachzeitschrift Nature Communications unter dem Titel „Unified differentable learning of electric response“ veröffentlicht.
Forschungshighlights:
* Das erste einheitliche Framework für maschinelles Lernen, das mehrdimensionale Erhaltungsgarantien für Impuls, elektrische Enthalpie usw. basierend auf der präzisen Differenzialbeziehung zwischen verallgemeinerter potenzieller Energie und beobachtbaren Reaktionsgrößen erreicht.
* Entwickeln Sie ein äquivariantes neuronales Netzwerkmodell, um den Engpass der ferroelektrischen Hysteresesimulation auf Millionenatomebene zu überwinden und die Domänennukleation und die eindimensionale Expansionsdynamik der Polarisationsumschaltung genau zu analysieren.
* Lösen Sie das Problem des mehrwertigen Polarisationstrainings und kombinieren Sie Grundprinzipien, um eine maßstabsübergreifende, hochpräzise Vorhersage der dielektrischen und ferroelektrischen Eigenschaften im α-SiO₂/BaTiO₃-System zu erreichen.

Papieradresse:
https://go.hyper.ai/18TWg
Weitere Artikel zu den Grenzen der KI:
α−SiO₂- und BaTiO₃-Datenexperimente, Aufbau des Trainingssatzes und Ableitung der Schlüsselparameter
In dieser Studie wurden Datenexperimente mit zwei Materialien durchgeführt: α-SiO₂ und BaTiO₃.Die Leistung des Modells wird durch die Erstellung von Trainings- und Validierungssätzen überprüft.
In der Phase der Trainingsdatengenerierung wurden 200 Frames der Atomkonfigurationen von α-SiO₂ aus klassischen NVT-MD-Simulationen bei 300 K und 600 K extrahiert (Vashishta-Potenzial-gesteuert, jeweils 100 ps lang, 1 ps Intervallabtastung). Für BaTiO₃ wurden 75 Frames (300 K–400 K, 60 Frames der Originalstruktur und 15 Frames der Domänenwandstruktur) durch aktive Lerndynamik des FLARE-Codes gesammelt.
Für jeden Datensatz wurden Energie, Kraft und Polarisation in Abwesenheit eines elektrischen Felds mittels DFT berechnet und die Bornsche Ladung und Polarisierbarkeit mithilfe der Methode der finiten Differenzen unter einem kleinen elektrischen Feld von 0,36 MV/cm abgeleitet, um den linearen Reaktionsbereich von Polarisation und elektrischem Feld sicherzustellen.
Rahmenwerk zur Vorhersage der Materialreaktion basierend auf einem äquivarianten neuronalen Netzwerk
In dieser Studie entwickelten Forscher erfolgreich ein innovatives Framework für maschinelles Lernen, das auf ein einheitliches Lernen verallgemeinerter potenzieller Energie- und Antwortfunktionen abzielt. Dieses Framework folgt strikt den mathematischen Prinzipien der Taylor-Reihe.Genialerweise werden die Lernaufgaben der verallgemeinerten potentiellen Energie und ihrer Ableitungen gleichzeitig innerhalb eines einheitlichen Modellrahmens ausgeführt.Dies ermöglicht eine genaue Vorhersage der Reaktionseigenschaften des Materials.
Das Kernkonzept dieses Modells besteht darin, dassDurch die Ableitung der verallgemeinerten potentiellen Energie in Bezug auf ihre Schlüsselvariablen (wie Atompositionen und externe Felder) können die entsprechenden Antworteigenschaften automatisch für jede Atomkonfiguration generiert werden.Es deckt viele Aspekte wie Kraft, Polarisation und Bornsche Ladung ab. Dieses Design gewährleistet nicht nur die genaue Umsetzung physikalischer Symmetrie und Erhaltungssätze wie Impulserhaltung und Erhaltung der elektrischen Enthalpie, sondern verbessert auch die Vorhersagegenauigkeit und Zuverlässigkeit des Modells erheblich.
In der Trainingsphase des ModellsDie Forscher erreichten dies, indem sie dem Input Parameter hinzufügten, die die Störungen des Systems beschreiben, wodurch das Modell diese Parameter unterscheiden konnte.Auf diese Weise kann das Training mit zusätzlichen physikalischen Größen durchgeführt werden. Gleichzeitig minimierten die Forscher eine umfassende Verlustfunktion, die die Beitragsfaktoren verschiedener Antworteigenschaften wie Energie, Kraft, Polarisation und Bornsche Ladung vollständig integriert. Dieser Trainingsmodus ist eng mit dem Konzept des Sobolev-Trainings verwandt. Genauer gesagt setzt sich jeder Term der Verlustfunktion aus der Differenz zwischen dem Gradienten, der dem Trainingslabel entspricht, und der Energie zusammen.
Um die nichtlineare Abhängigkeit der Antworteigenschaften von mehreren Feldern und die Kopplungseigenschaften genau zu erfassen,Die Forscher verwendeten eine neuronale Netzwerkarchitektur und eine leistungsstarke Technik namens Gradient Backpropagation.Lernen Sie das komplexe Beziehungsnetzwerk zwischen verallgemeinerten Potenzialen und ihren Eingaben gründlich kennen.
Beim Aufbau der ModellarchitekturForscher verlassen sich auf die Allegro-Plattform.Durch die volle Ausnutzung der hohen Präzision und herausragenden Dateneffizienz äquivarianter neuronaler NetzwerkeGleichzeitig erreichte das Modell durch die strikte Einhaltung des lokalen Designkonzepts eine hervorragende Skalierbarkeit. Im Hinblick auf das Design der Modelleingaben, wie in der Abbildung dargestellt, haben die Forscher das elektrische Feld E geschickt mit der Atomposition r_i zusammengeführt, wodurch das Modell die Lernaufgaben der elektrischen Enthalpie U, der Kraft F_i, der Polarisation P, der Born-Ladung Z_i und der Polarisierbarkeit α gleichzeitig ausführen kann.
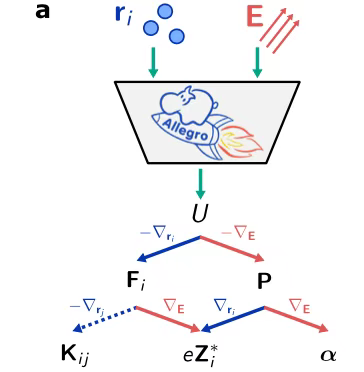
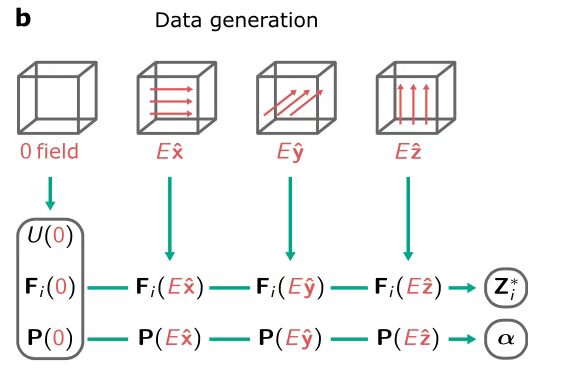
Im Prozess der Generierung von TrainingsdatenWie in der folgenden Abbildung dargestellt, verwendeten die Forscher DFT-Berechnungen, um Daten im Bereich um das elektrische Nullfeld zu erhalten, und nutzten die Methode der finiten Differenzennäherung, um die Werte der Bornschen Ladung und Polarisierbarkeit zu bestimmen. Nach dem Training kann das Modell Enthalpie ausgeben und anschließend eine Reihe wichtiger Parameter wie Kraft, Polarisation, Bornsche Ladung und Polarisierbarkeit ableiten, indem die Ableitungen erster und zweiter Ordnung der Ausgabeenthalpie berechnet werden.
Erwähnenswert ist, dass die Forscher speziell eine entsprechende Schnittstelle entwickelt haben, damit das Modell nahtlos in die LAMMPS-Software integriert werden kann und so groß angelegte Strukturrelaxations- und maschinelle Lern-Molekulardynamik-Simulationen (MLMD) unter elektrischen Feldbedingungen stark unterstützt werden.
Überprüfung der Modellgenauigkeit anhand mehrerer Szenarien
Um die Leistung des Modells zu überprüfen, wurden im Rahmen der Forschung Experimente in zwei Hauptrichtungen durchgeführt: Materialschwingung und dielektrische Eigenschaften, ferroelektrische Hysterese und Dipoldynamik.Anhand von α-SiO₂ und BaTiO₃ als Forschungsobjekte werden die Genauigkeit und Anwendbarkeit des Modells in verschiedenen Szenarien eingehend untersucht.
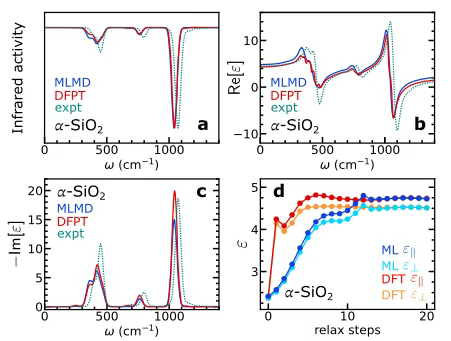
Bei der Untersuchung von Schwingungs- und dielektrischen EigenschaftenDie Forscher wählten α-SiO₂ als typisches Forschungsobjekt, extrahierten 200 Datenframes mit 72 Atomen aus der Molekulardynamiksimulation der klassischen potentiellen Energie und trainierten darauf basierend das entsprechende maschinelle Lernmodell. Um die Genauigkeit des Modells weiter zu untersuchen, wurde eine Superzelle mit 24.696 Atomen konstruiert. Nach 10 Pikosekunden Gleichgewicht unter dem NVE-Ensemble wurde eine 200 Pikosekunden lange maschinelle Lern-Molekulardynamiksimulation (MLMD) ohne elektrisches Feld durchgeführt. Das Infrarotspektrum wurde mithilfe der Polarisationsdynamik berechnet und die Polarisations- und Polarisierbarkeitsdynamik analysiert, um die frequenzabhängige Dielektrizitätskonstante zu bestimmen. Schließlich wurden die Ergebnisse mit den Berechnungsergebnissen basierend auf der Dichtefunktionalstörungstheorie (DFPT) und experimentellen Daten verglichen.
Wie in Abbildung ac unten gezeigt,Die Ergebnisse von MLMD und DFPT waren sehr konsistent.Zur Untersuchung der Abschirmwirkung von Elektronen und Ionen in Gegenwart eines elektrischen Felds führten die Forscher zudem eine Strukturrelaxation unter einem begrenzten elektrischen Feld basierend auf der ursprünglichen Volumenstruktur von α-SiO₂ durch. Die statische Dielektrizitätskonstante wurde durch Berechnung der Polarisationsdifferenz zwischen dem elektrischen Feld und dem Zustand ohne elektrisches Feld bestimmt. Die Ergebnisse sind in Abbildung d unten dargestellt. Die mit dem Modell ermittelten hochfrequenten und statischen Dielektrizitätskonstanten stimmen grundsätzlich mit den DFT-Werten überein.Dies zeigt deutlich, dass das Modell den Beitrag des elektrischen Felds zur elektronischen Struktur genau erfassen kann, und bestätigt seine Wirksamkeit in Simulationen der Dynamik endlicher elektrischer Felder.
Gleichzeitig wurde bei der Trainingsüberprüfung der Born-Ladungen festgestellt, dass die Genauigkeit der Schwingung und der dielektrischen Reaktion bei niedrigen Frequenzen erheblich beeinträchtigt wird, wenn das Modell nicht mit Born-Ladungen trainiert wird, insbesondere bei begrenzten Daten, und dass der Rechenaufwand der Simulation unter elektrischen Feldern steigt.Dieses Ergebnis unterstreicht, dass das Training mit Born-Ladungen ein entscheidender Vorteil des Modells ist.
Bei der Untersuchung der ferroelektrischen Hysterese und DipoldynamikDie Forscher konzentrierten sich auf BaTiO₃-Perowskit und nutzten 75 Datenframes, die durch aktive Lerndynamik mit 135 Atomen pro Frame extrahiert wurden, um das maschinelle Lernmodell zu trainieren. Die ferroelektrische Hysterese der 135-Atom-Superzelle wurde bei Nulltemperatur berechnet. Die Ergebnisse sind in Abbildung a unten dargestellt.Die durch die MLMD-Simulation ermittelte ferroelektrische Hysterese stimmt weitgehend mit der durch die DFT-Berechnung ermittelten überein, was die Zuverlässigkeit des Modells deutlich bestätigt.
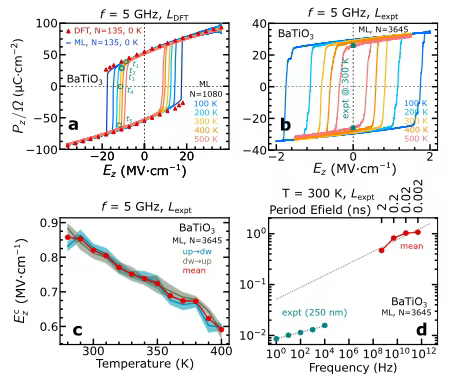
Um den Einfluss der Temperatur auf die ferroelektrische Reaktion weiter zu untersuchen, führten die Forscher eine MLMD im NVT-Ensemble unter einem sinusförmigen elektrischen Feld von 5 GHz durch. Die Studie ergab, dass, wie in den Abbildungen bc oben dargestellt,Mit steigender Temperatur nimmt das intrinsische Koerzitivfeld ab, während die spontane Polarisation relativ weniger von der Temperatur beeinflusst wird.Sowohl bei Nulltemperatur als auch bei endlicher Temperatur zeigen die Hysteresekurven eine Symmetrie des elektrischen Feldes, was mit den Eigenschaften polarisationskonservativer Vektorfelder übereinstimmt. Darüber hinaus stimmt die spontane Polarisation bei der Verwendung einer 3.645-atomigen Superzelle mit experimentellen Gitterparametern für Forschungszwecke mit den experimentellen Ergebnissen überein, und auch das Koerzitivfeld liegt näher am experimentellen Wert. Dies bestätigt die Fähigkeit des Modells zur Extrapolation unter Superzellen mit unterschiedlichen Ordnungszahlen und Gitterparametern.
Bei der Untersuchung der Auswirkung der Frequenz des elektrischen Feldes auf die ferroelektrische HystereseDie MLMD einer 3.645-atomigen Superzelle bei 300 K zeigt, dass das Koerzitivfeld mit abnehmender Frequenz abnimmt. Obwohl der extrapolierte Wert immer noch etwa eine Größenordnung vom experimentellen Wert abweicht, unterstreicht diese komplexe Rechenaufgabe die hohe Rechenleistung des Modells und seine hervorragende Skalierbarkeit auf millionenatomige Systeme.
Materialmodellforschung: Gemeinsame Fortschritte zwischen Wissenschaft und Industrie
Im Bereich der Materialwissenschaften haben sowohl die Hochschulen als auch die Wirtschaft große Anstrengungen unternommen, um die Weiterentwicklung der Forschung im Bereich Materialmodelle voranzutreiben.
Viele Universitäten und Forschungsteams haben in der akademischen Gemeinschaft bemerkenswerte Ergebnisse erzielt. Forscher der University of Rochester in den USA haben ein maschinelles Lernmodell entwickelt, das die enormen Datenmengen aus Röntgenbeugungsexperimenten (XRD) analysieren und so die Materialinnovation beschleunigen kann. Das Modell wird mit experimentellen Daten anorganischer Materialien unter verschiedenen Versuchsbedingungen und Kristalleigenschaften trainiert und gemäß dem Bragg-Gesetz klassifiziert und optimiert. Das vom Imperial College of London vorgeschlagene Modell Chemeleon nutzt generative KI, um anhand des Datensatzes mit den Strukturmerkmalen des Materials zu navigieren, aus Textbeschreibungen und dreidimensionalen Strukturdaten zu lernen und Stichproben der chemischen Zusammensetzung und Kristallstruktur zu gewinnen. Das Team der Seoul National University in Südkorea und der Fordham University in den USA verwendete ein großes Sprachmodell (LLM), um die Synthetisierbarkeit neuer Materialien vorherzusagen, und erläuterte die Grundlagen der Vorhersage, wobei es gute Leistung und Interpretierbarkeit zeigte. Das Team der Professoren Wang Jinlan und Ma Liang von der School of Physics der Southeast University in China und das Team von Professor Wang Xinran von der Nanjing University schlugen neue Ideen in der Erforschung zweidimensionaler Materialien vor, realisierten das zahlenkontrollierte epitaktische Wachstum von gleichmäßigen doppelschichtigen MoS₂-Dünnfilmen im Zentimeterbereich und schlugen Methoden zur Verbesserung der Leistung von Kontaktschnittstellen durch Computersimulation vor, wodurch erfolgreich ein ultraniedriger Kontaktwiderstand erreicht wurde.
Die Wirtschaft steht dem in nichts nach und engagiert sich aktiv für die Innovation und Anwendung von Materialmodelltechnologie. Apple nutzt maschinelle Lernmodelle, um Metalllegierungen für die Herstellung von Produktschalen zu optimieren, die Materialfestigkeit und Haltbarkeit zu verbessern und Kosten zu senken. BASF entwickelt fortschrittliche Materialmodelle, um Materialeigenschaften zu simulieren, die Entwicklung neuer Kunststoffe und Beschichtungen zu beschleunigen und die Wettbewerbsfähigkeit von Produkten zu verbessern. Das von Microsoft eingeführte Modell MatterGen generiert Materialstrukturen, die den Designanforderungen durch eine einzigartige Diffusionsmodellarchitektur entsprechen und gegenüber herkömmlichen Methoden erhebliche Vorteile bieten. Darüber hinaus hat das Unternehmen mit dem SIAT-Team der Chinesischen Akademie der Wissenschaften zusammengearbeitet, um das neue Material TaCr₂O₆ zu entwickeln.
Diese hochmodernen Forschungen und innovativen Verfahren greifen ineinander und treiben die Materialmodellforschung kontinuierlich voran. Mit der fortschreitenden Vertiefung der Forschung und der Weiterentwicklung und Verbesserung der Technologie werden Materialmodelle in Zukunft Durchbrüche und Anwendungen in einem breiteren Spektrum von Bereichen erzielen und so eine solide Grundlage für die wissenschaftliche und technologische Entwicklung schaffen.
Referenzartikel:
1.https://mp.weixin.qq.com/s/mctu0DOWO_OieLnOgp93Rw
2.https://mp.weixin.qq.com/s/I-UZTyUFSWwXlf1LCmwjRQ
3.https://mp.weixin.qq.com/s/Ox62ut3IJcUWsLC7sF100Q
4.https://mp.weixin.qq.com/s/VlPb8zSghVVxnPNl-WzqBA
5.https://plastics-rubber.basf.com/global/en/performance_polymers/services/service_ultrasim/Material-Modeling








